tests
A repository to share problems under development.
1. AI-related materials
- Water Margin riddles (《水浒》谜语). https://jinghuazhao.github.io/tests/AI/.
- Supplementary materials in Python. https://jinghuazhao.github.io/tests/AI/python.
2. METAL
https://jinghuazhao.github.io/tests/METAL/
3. turboqq & turboman
- turboqq for https://github.com/bpprins/turboqq
- turboman for https://github.com/bpprins/turboman
> options(width=200) > load("turboman_hg19_reference_data.rda") > ls() [1] "ld_block_breaks_pickrell_hg19_eur" "refgene_gene_coordinates_h19" > head(ld_block_breaks_pickrell_hg19_eur) chr start 1 1 10583 2 1 1892607 3 1 3582736 4 1 4380811 5 1 5913893 6 1 7247335 > head(refgene_gene_coordinates_h19) chromosome gene_transcription_start gene_transcription_stop gene_name gene_transcription_midposition 1 1 11873 14409 DDX11L1 13141.0 53009 1 17368 17436 MIR6859-1 17402.0 55877 1 17368 17436 MIR6859-3 17402.0 2 1 14361 29370 WASH7P 21865.5 45554 1 30365 30503 MIR1302-10 30434.0 45166 1 30365 30503 MIR1302-9 30434.0 - Annotation files which be can used to build
turboman_hg19_reference_data.rda.- glist-hg19 and glist-hg38, https://www.cog-genomics.org/plink/1.9/resources
- ldetect-data, https://bitbucket.org/nygcresearch/ldetect-data/src/master/
- The IL-12B example,
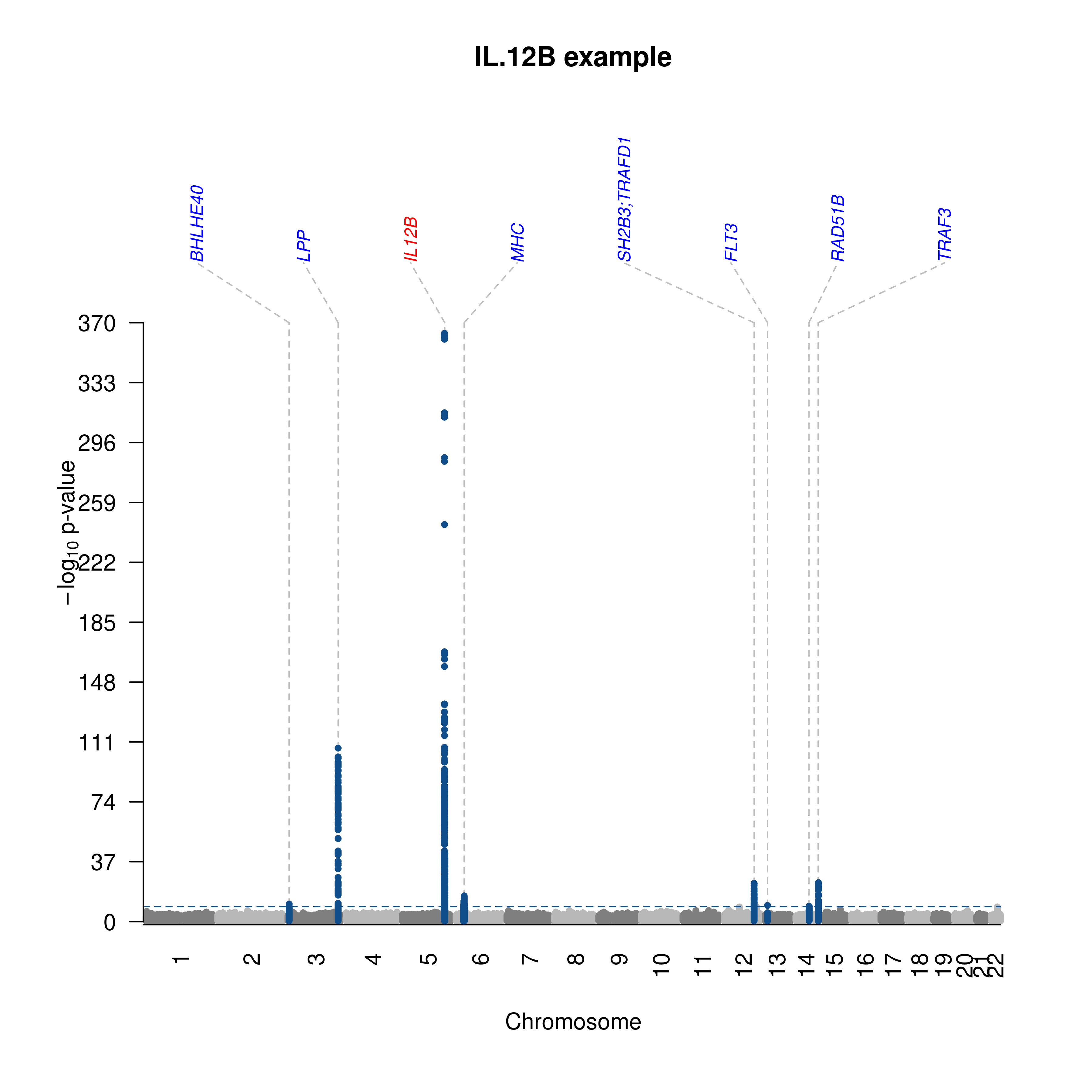
- IL.12B.txt.gz was split via
split --bytes=45M "IL.12B.txt.gz" "IL.12B.txt-" - It could be recoverd via
cat IL.12B.txt-*> IL.12B.txt.gz
- IL.12B.txt.gz was split via
4. SNPid
A SNPid (chr:pos_a1/a2) often replaces RSid as an unique variant identifier in genetic association studies. A customised implemention for a compressed VCF file based on Bash is as follows,
#!/usr/bin/bash
function snpid2()
{
gunzip -c ${1} | \
awk -v use_I_D_as_alleles=0 -v FS="\t" -v OFS="\t" -v out=${2} '
NR==1,/#CHROM/{print;next}
{
if (length($4)>1||length($5)>1) {if (length($4)>length($5)) {a1="I"; a2="D"} else {a1="D"; a2="I"}; $3=$1":"$2"_D/I"} else
{a1=$4; a2=$5; if (a1<a2) $3=$1":"$2"_"a1"/"a2; else $3=$1":"$2"_"a2"/"a1}
n=a[$3]++
if(n>0) {a1=a1 n; a2=a2 n; $3=$1":"$2"_"a1"/"a2 }
if(use_I_D_as_alleles) {$4=a1; $5=a2}
if(length($4)>1||length($5)>1) print $1,$2,$3,$4,$5 >> sprintf("%s.txt",out)
print
}' | bgzip -f > ${2}-snpid.vcf.gz
bcftools index -tf ${2}-snpid.vcf.gz
}
rm -f test.txt
snpid2 ERZ127238/HPSI1013i-garx_3.wec.gtarray.HumanCoreExome-12_v1_0.imputed_phased.20150604.genotypes.vcf.gz test
Only definitions for indels are listed. More details are available from the snpid directory.