1 LocusZoom.js plots
1.1 A list of plots
This figure starts with a default and is updated when selected for one of the two alternatives.
htmltools::tags$iframe(src = "/pQTLtools/articles/lz.html", width = "100%", height = "600px")THe HTML skeleton (lz.html) takes a file named top_hits.json containing the menu items,
[
["A1BG", "rs145685027", "19:58948122"],
["ACE", "rs4353", "17:61570422"]
]each corresponds to a file for GWAS summary statistics as shown below
> d <- jsonlite::fromJSON("vignettes/data/ACE-rs4353.json")
> class(d)
[1] "list"
> lapply(d,class)
$ppid
[1] "character"
$data
[1] "data.frame"
> lapply(d,head,3)
$ppid
[1] "ACE-rs4353"
$data
variant position ref_allele alt_allele_freq beta log_pvalue
1 17:61322094_G/T 61322094 G 0.0154 -0.1899 0.69
2 17:61322221_G/A 61322221 G 0.1905 0.0190 0.22
3 17:61322307_G/T 61322307 G 0.9682 0.4065 5.65
> j <- jsonlite::toJSON(lapply(d,head,3))
> j
{"ppid":["ACE-rs4353"],"data":[{"variant":"17:61322094_G/T","position":61322094,"ref_allele":"G","alt_allele_freq":0.0154,"beta":-0.1899,"log_pvalue":0.69},{"variant":"17:61322221_G/A","position":61322221,"ref_allele":"G","alt_allele_freq":0.1905,"beta":0.019,"log_pvalue":0.22},{"variant":"17:61322307_G/T","position":61322307,"ref_allele":"G","alt_allele_freq":0.9682,"beta":0.4065,"log_pvalue":5.65}]}1.2 Stacked association plots
The figure below extends to three panels.
htmltools::tags$iframe(src = "/pQTLtools/articles/stack.html", width = "100%", height = "600px")2 gap::asplot
This section mirrors the original CDKN results for the named function.
pkgs <- c("AnnotationDbi", "GenomicFeatures", "GenomicRanges", "IRanges", "TxDb.Hsapiens.UCSC.hg19.knownGene",
"bigsnpr", "ieugwasr", "zoo", "org.Hs.eg.db")
for (p in pkgs) if (length(grep(paste("^package:", p, "$", sep=""), search())) == 0) {
if (!requireNamespace(p)) warning(paste0("This vignette needs package `", p, "'; please install"))
}
#> Loading required namespace: AnnotationDbi
#> Loading required namespace: GenomicFeatures
#> Loading required namespace: TxDb.Hsapiens.UCSC.hg19.knownGene
#> Loading required namespace: bigsnpr
#> Loading required namespace: ieugwasr
#> Loading required namespace: zoo
#> Loading required namespace: org.Hs.eg.db
#>
invisible(suppressMessages(lapply(pkgs, require, character.only = TRUE)))
chr <- 9
start <- 21900000
end <- 22500000
opengwas_id <- "ebi-a-GCST006867"
region <- paste0(chr, ":", start, "-", end)
dat <- ieugwasr::associations(variants=region, id=opengwas_id)
#> Querying id chunk 1 of 1
#> Querying variant chunk 1 of 1
lead <- dat$rsid[which.min(dat$p)]
dat <- dat[order(dat$p), ]
snps500 <- unique(dat$rsid)
snps500 <- na.omit(snps500)
snps500 <- snps500[1:min(500, length(snps500))]
ld_mat <- ieugwasr::ld_matrix(variants=snps500, pop="EUR")
#> Please look at vignettes for options on running this locally if you need to run many instances of this command.
ld_names <- rownames(ld_mat)
ld_clean_names <- sub("_.*", "", ld_names)
rownames(ld_mat) <- ld_clean_names
colnames(ld_mat) <- ld_clean_names
dat_gap <- data.frame(chr=paste0("chr", dat$chr), pos=dat$position, snp=dat$rsid)
dat_gap$logp <- -log10(dat_gap$p)
stopifnot(all(rownames(ld_mat)==colnames(ld_mat)))
rsqr <- ld_mat[lead, ]^2
dat$rsqr <- rsqr[match(dat$rsid, names(rsqr))]
dat$gpos_cM <- bigsnpr::snp_asGeneticPos(infos.chr=dat$chr, infos.pos=dat$position, type="OMNI")
dat <- dat[order(dat$chr, dat$position), ]
dat$d_cm <- c(NA, diff(dat$gpos_cM))
dat$theta <- c(NA, (1 - exp(-2 * dat$d_cm[-1] / 100)) / 2)
locus <- data.frame(CHR=dat$chr, POS=dat$position, NAME=dat$rsid, PVAL=dat$p, RSQR=dat$rsqr)
stopifnot(all(is.finite(locus$PVAL)))
stopifnot(all(locus$PVAL >= 0 & locus$PVAL <= 1))
locus <- locus[!is.na(locus$CHR) & !is.na(locus$POS), ]
if (nrow(locus) == 0) {stop("ERROR: locus is empty after cleaning dat")}
locus <- locus[order(locus$CHR, locus$POS), ]
locus$CM <- snp_asGeneticPos(infos.chr=locus$CHR, infos.pos=locus$POS, type="OMNI")
locus$RATE <- NA_real_
ok <- !is.na(locus$CM)
if (sum(ok) > 1) {locus$RATE[ok] <- c(NA, diff(locus$CM[ok]) / diff(locus$POS[ok]) * 1e6)}
if (sum(!is.na(locus$RATE))>3) {locus$RATE <- zoo::rollmean(locus$RATE, k=7, fill=NA, align="center")}
stopifnot(nrow(locus) > 0)
stopifnot(all(is.na(locus$RATE) | is.finite(locus$RATE)))
stopifnot(all(is.na(locus$CM) | locus$CM>=0))
stopifnot(all(c("POS", "CM", "RATE") %in% colnames(locus)))
locus <- locus[order(locus$POS), ]
map <- with(locus,data.frame(POS=POS, THETA=RATE, DIST=CM))
map <- map[order(map$POS), ]
map$THETA[!is.finite(map$THETA)] <- NA
map$DIST[!is.finite(map$DIST)] <- NA
gr <- GenomicRanges::GRanges(seqnames=paste0("chr", chr),ranges=IRanges::IRanges(start, end))
genes_gr <- GenomicFeatures::genes(TxDb.Hsapiens.UCSC.hg19.knownGene::TxDb.Hsapiens.UCSC.hg19.knownGene)
#> 24 genes were dropped because they have exons located on both strands of the
#> same reference sequence or on more than one reference sequence, so cannot be
#> represented by a single genomic range.
#> Use 'single.strand.genes.only=FALSE' to get all the genes in a GRangesList
#> object, or use suppressMessages() to suppress this message.
hits <- IRanges::subsetByOverlaps(genes_gr, gr)
symbols <- AnnotationDbi::mapIds(org.Hs.eg.db::org.Hs.eg.db, keys=hits$gene_id, keytype="ENTREZID", column="SYMBOL")
#> 'select()' returned 1:1 mapping between keys and columns
genes_df <- data.frame(gene_id=hits$gene_id, symbol=S4Vectors::unname(symbols), start=GenomicRanges::start(hits),
end=GenomicRanges::end(hits), strand=as.character(GenomicRanges::strand(hits)))
genes <- data.frame(START=genes_df$start, STOP=genes_df$end, STRAND=genes_df$strand, GENE=genes_df$symbol)
gap::asplot(locus,map,genes)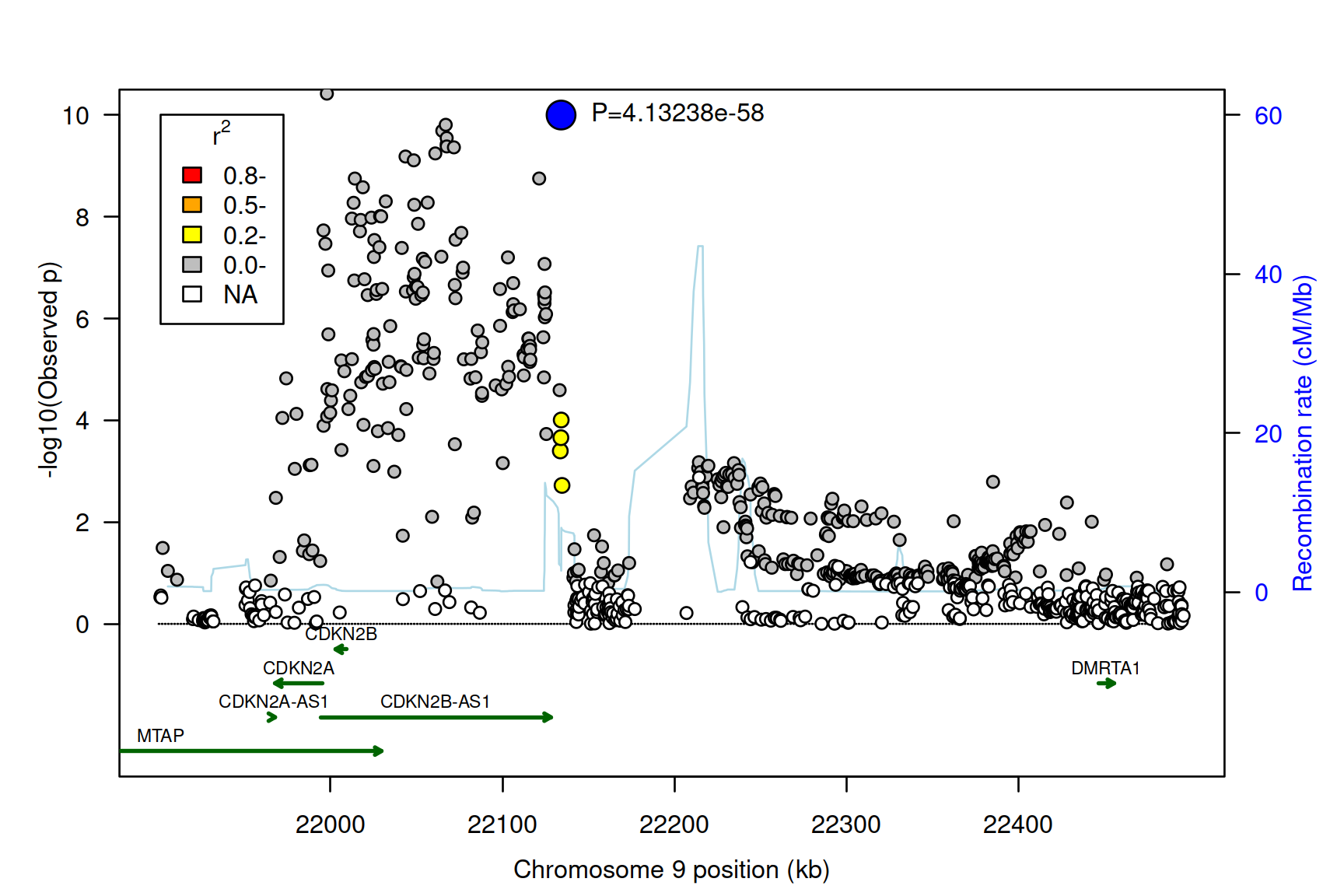
Figure 2.1: Association plot for T2D
3 SCALLOP analysis
This section details colocalization and pQTL/disease overlap analysis.
3.1 Colocalization
This is the actual script for cis-pQTL colocalization analysis on GTEx v8 for SCALLOP-INF.
3.1.1 Data
The data were GWAS summary statistics in GRCh37 and VCF format, converted by gwasvcf. The GTEx association statistics were in GRCh38 and downloaded from the eQTL Catalogue and stored locally. Data on microarray and RNA-Seq remain on the eQTL Catalogue website.
3.1.2 coloc.R
It contains minor modification to the documentation example,
liftRegion <- function(x,chain,flanking=1e6)
{
require(GenomicRanges)
gr <- with(x,GenomicRanges::GRanges(seqnames=chr,IRanges::IRanges(start,end))+flanking)
seqlevelsStyle(gr) <- "UCSC"
gr38 <- rtracklayer::liftOver(gr, chain)
chr <- gsub("chr","",colnames(table(seqnames(gr38))))
start <- min(unlist(start(gr38)))
end <- max(unlist(end(gr38)))
invisible(list(chr=chr[1],start=start,end=end,region=paste0(chr,":",start,"-",end)))
}
sumstats <- function(prot,chr,region37)
{
cat("GWAS sumstats\n")
vcf <- file.path(INF,"METAL/gwas2vcf",paste0(prot,".vcf.gz"))
gwas_stats <- gwasvcf::query_gwas(vcf, chrompos = region37) %>%
gwasvcf::vcf_to_granges() %>%
keepSeqlevels(chr) %>%
renameSeqlevels(paste0("chr",chr))
gwas_stats_hg38 <- rtracklayer::liftOver(gwas_stats, chain) %>%
unlist() %>%
# renameSeqlevels(chr) %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames,
position = start, REF, ALT, AF, ES, SE, LP, SS) %>%
dplyr::mutate(id = paste(chromosome, position, sep = ":")) %>%
dplyr::mutate(MAF = pmin(AF, 1-AF)) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub("chr","",chromosome))
}
microarray <- function(gwas_stats_hg38,ensGene,region38)
{
cat("a. eQTL datasets\n")
microarray_df <- dplyr::filter(tabix_paths, quant_method == "microarray") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(microarray_df$ftp_path), microarray_df$qtl_id[1])
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.CEDAR")
column_names <- names(read.delim(hdr))
summary_list <- purrr::map(ftp_path_list, ~import_eQTLCatalogue(., region38,
selected_gene_id = ensGene, column_names))
purrr::map_df(summary_list[lapply(summary_list,nrow)!=0],
~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
rnaseq <- function(gwas_stats_hg38,ensGene,region38)
{
cat("b. Uniformly processed RNA-seq datasets\n")
rnaseq_df <- dplyr::filter(tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(rnaseq_df$ftp_path), rnaseq_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list, ~safe_import(., region38,
selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list[lapply(result_list,nrow)!=0], ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
purrr::map_df(result_list, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
gtex <- function(gwas_stats_hg38,ensGene,region38)
{
cat("c. GTEx_v8 imported eQTL datasets\n")
gtex_df <- dplyr::filter(imported_tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(gtex_df$ftp_path), gtex_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.GTEx")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
coloc <- function(prot,chr,ensGene,chain,region37,region38,out,run_all=FALSE)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
if (exists("df_gtex"))
{
saveRDS(df_gtex,file=paste0(out,".RDS"))
dplyr::arrange(df_gtex, -PP.H4.abf)
p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram()
}
if (run_all)
{
df_microarray <- microarray(gwas_stats_hg38,ensGene,region38)
df_rnaseq <- rnaseq(gwas_stats_hg38,ensGene,region38)
if (exists("df_microarray") & exits("df_rnaseq") & exists("df_gtex"))
{
coloc_df = dplyr::bind_rows(df_microarray, df_rnaseq, df_gtex)
saveRDS(coloc_df, file=paste0(out,".RDS"))
dplyr::arrange(coloc_df, -PP.H4.abf)
p <- ggplot(coloc_df, aes(x = PP.H4.abf)) + geom_histogram()
}
}
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, "-assoc.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, "-hist.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
single_run <- function(r)
{
sentinel <- sentinels[r,]
chr <- with(sentinel,Chr)
ss <- subset(inf1,prot==sentinel[["prot"]])
ensRegion37 <- with(ss,
{
start <- start-M
if (start<0) start <- 0
end <- end+M
paste0(chr,":",start,"-",end)
})
ensGene <- ss[["ensembl_gene_id"]]
ensRegion38 <- with(liftRegion(ss,chain),region)
f <- file.path(INF,"coloc",with(sentinel,paste0(prot,"-",SNP)))
cat(chr,ensGene,ensRegion37,ensRegion38,"\n")
coloc(sentinel[["prot"]],chr,ensGene,chain,ensRegion37,ensRegion38,f)
}
# slow with the following loop:
loop <- function() for (r in 1:nrow(sentinels)) single_run(r)
library(pQTLtools)
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
pkgs <- c("dplyr", "ggplot2", "readr", "coloc", "GenomicRanges","seqminer")
invisible(lapply(pkgs, require, character.only = TRUE))
HPC_WORK <- Sys.getenv("HPC_WORK")
gwasvcf::set_bcftools(file.path(HPC_WORK,"bin","bcftools"))
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths.tsv")
tabix_paths <- read.delim(f, stringsAsFactors = FALSE) %>% dplyr::as_tibble()
HOME <- Sys.getenv("HOME")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_gtex.tsv")
imported_tabix_paths <- within(read.delim(fp, stringsAsFactors = FALSE) %>% dplyr::as_tibble(),
{
f <- lapply(strsplit(ftp_path,"/csv/|/ge/"),"[",3)
ftp_path <- paste0("~/rds/public_databases/GTEx/csv"),f)
})
library(dplyr)
INF <- Sys.getenv("INF")
M <- 1e6
sentinels <- subset(read.csv(file.path(INF,"work","INF1.merge.cis.vs.trans")),cis)
cvt_rsid <- file.path(INF,"work","INF1.merge.cis.vs.trans-rsid")
prot_rsid <- subset(read.delim(cvt_rsid,sep=" "),cis,select=c(prot,SNP))
# Faster with parallel Bash runs.
r <- as.integer(Sys.getenv("r"))
single_run(r)where options for protein GWAS, microarray, RNA-Seq are available with respect to variant-flanking or gene regions. When no results are generated,
there would have problem with dplyr::arrange(df_gtex, -PP.H4.abf);p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram().
3.1.3 Collection of results
When these are furnished we keep results (i.e., PP4>=0.8) as follows,
collect <- function()
{
df_coloc <- data.frame()
for(r in 1:nrow(sentinels))
{
prot <- sentinels[["prot"]][r]
snpid <- sentinels[["SNP"]][r]
rsid <- prot_rsid[["SNP"]][r]
f <- file.path(INF,"coloc",paste0(prot,"-",snpid,".RDS"))
if (!file.exists(f)) next
cat(prot,"-",rsid,"\n")
rds <- readRDS(f)
if (nrow(rds)==0) next
df_coloc <- rbind(df_coloc,data.frame(prot=prot,rsid=rsid,snpid=snpid,rds))
}
df_coloc <- within(df_coloc,{qtl_id <- gsub("GTEx_V8_","",qtl_id)}) %>%
rename(H0=PP.H0.abf,H1=PP.H1.abf,H2=PP.H2.abf,H3=PP.H3.abf,H4=PP.H4.abf)
write.table(subset(df_coloc,H4>=0.8),
file=file.path(INF,"coloc","GTEx.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
}
collect()3.1.4 The driver program
It is in Bash.
#!/usr/bin/bash
for r in {1..59}
do
export r=${r}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
done3.1.5 Parallel computing
To speed up the analysis, we resort to SLURM,
#!/usr/bin/bash
#SBATCH --job-name=_coloc
#SBATCH --account CARDIO-SL0-CPU
#SBATCH --partition cardio
#SBATCH --qos=cardio
#SBATCH --array=1-59
#SBATCH --mem=28800
#SBATCH --time=5-00:00:00
#SBATCH --error=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.err
#SBATCH --output=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.out
#SBATCH --export ALL
export trait=$(awk 'NR==ENVIRON["SLURM_ARRAY_TASK_ID"] {print $1}' ${INF}/work/inf1.tmp)
function gtex()
{
export r=${SLURM_ARRAY_TASK_ID}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
}
gtex3.2 pQTL/disease overlap
The ontology of traits/disease is available through Experimental Factor Ontology (EFO)1, which can be used to build lists of diseases and immune-mediated traits and filter search results from PhenoScanner2.
3.2.1 Diseases
library(ontologyIndex)
# http://www.ebi.ac.uk/efo/efo.obo
INF <- Sys.getenv("INF")
file <- file.path(INF,"ebi","efo-3.26.0","efo.obo")
get_relation_names(file)
efo <- get_ontology(file, extract_tags="everything")
id <- function(ontology)
{
length(ontology)
length(ontology$id)
inf <- grep(ontology$name,pattern="immune|inflammatory")
data.frame(id=ontology$id[inf],name=ontology$name[inf])
}
goidname <- id(go)
efoidname <- id(efo)
# all diseases
efo_diseases <- get_descendants(efo,"EFO:0000408")
diseases_name <- efo$name[efo_diseases]
diseases <- data.frame(efo_diseases,diseases_name)
write.table(diseases,file=file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=FALSE,row.names=FALSE,sep=",")
# immune system diseases (isd)
efo_0000540 <- get_descendants(efo,"EFO:0000540")
efo_0000540name <- efo$name[efo_0000540]
isd <- data.frame(efo_0000540,efo_0000540name)
library(ontologyPlot)
onto_plot(efo,efo_0000540)3.2.2 Lookup
suppressMessages(library(dplyr))
suppressMessages(library(gap))
suppressMessages(library(pQTLtools))
inf1_prot <- vector()
for(i in 1:92) inf1_prot[inf1[i,"prot"]] <- mutate(inf1[i,],target.short=if_else(!is.na(alt_name),alt_name,target.short))[["target.short"]]
INF1_metal <- within(read.delim(file.path(find.package("pQTLtools"),"tests","INF1.METAL"),as.is=TRUE),{
hg19_coordinates=paste0("chr",Chromosome,":",Position)}) %>%
rename(INF1_rsid=rsid, Total=N) %>%
left_join(pQTLdata::inf1[c("prot","gene","target.short","alt_name")]) %>%
mutate(target.short=if_else(!is.na(alt_name),alt_name,target.short)) %>%
select(-alt_name)
INF1_aggr <- INF1_metal %>%
select(Chromosome,Position,target.short,gene,hg19_coordinates,
MarkerName,Allele1,Allele2,Freq1,Effect,StdErr,log.P.,cis.trans,INF1_rsid) %>%
group_by(Chromosome,Position,MarkerName,INF1_rsid,hg19_coordinates) %>%
summarise(nprots=n(),
prots=paste(target.short,collapse=";"),
Allele1=paste(toupper(Allele1),collapse=";"),
Allele2=paste(toupper(Allele2),collapse=";"),
EAF=paste(Freq1,collapse=";"),
Effects=paste(Effect,collapse=";"),
SEs=paste(StdErr,collapse=";"),
log10P=paste(log.P.,collapse=";"),
cistrans=paste(cis.trans,collapse=";")) %>%
data.frame()
rsid <- INF1_aggr[["INF1_rsid"]]
catalogue <- "GWAS"
proxies <- "EUR"
p <- 5e-8
r2 <- 0.8
build <- 37
INF <- Sys.getenv("INF")
efo_diseases <- read.table(file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=c("efo","disease"),as.is=TRUE,sep=",") %>%
mutate(efo=gsub(":", "_", efo))
r <- snpqueries(rsid, catalogue=catalogue, proxies=proxies, p=p, r2=r2, build=build)
lapply(r,dim)
snps_results <- with(r,right_join(snps,results))
ps <- subset(snps_results,select=-c(hg38_coordinates,ref_hg38_coordinates,pos_hg38,ref_pos_hg38,dprime))
aggr <- subset(within(INF1_aggr,{HLA <- as.numeric(Chromosome==6 & Position >= 25392021 & Position <= 33392022)}),
select=-c(Chromosome,Position,INF1_rsid))
short <- merge(aggr,ps,by="hg19_coordinates")
gwas <- function()
{
short <- merge(aggr,ps,by="hg19_coordinates") %>%
filter(efo %in% pull(efo_diseases,efo)) %>%
left_join(efo_diseases)
v <- c("prots","hgnc","MarkerName","cistrans","Effects","Allele1","Allele2","rsid","a1","a2","efo",
"ref_rsid","ref_a1","ref_a2","proxy","r2",
"HLA","beta","se","p","disease","n_cases","n_controls","unit","ancestry","pmid","study")
mat <- within(short[v],
{
flag <- (HLA==1)
prefix <- paste0(prots,"-",rsid)
prefix[flag] <- paste0(prefix[flag],"*")
rsidProts <- paste0(prefix," (",hgnc,")")
efoTraits <- gsub("\\b(^[a-z])","\\U\\1",disease,perl=TRUE)
qtl_direction <- sign(as.numeric(beta))
})
combined <- group_by(mat,efoTraits,rsidProts,desc(n_cases)) %>%
summarize(direction=paste(qtl_direction,collapse=";"),
betas=paste(beta,collapse=";"),
units=paste(unit,collapse=";"),
studies=paste(study,collapse=";"),
diseases=paste(disease,collapse=";"),
cases=paste(n_cases,collapse=";")
) %>%
data.frame()
rxc <- with(combined,table(efoTraits,rsidProts))
for(cn in colnames(rxc)) for(rn in rownames(rxc)) {
cnrn <- subset(combined,efoTraits==rn & rsidProts==cn)
if(nrow(cnrn)==0) next
rxc[rn,cn] <- as.numeric(unlist(strsplit(cnrn[["direction"]],";"))[1])
}
write.table(mat,file=file.path(INF,"work","pQTL-disease-GWAS.csv"),row.names=FALSE,quote=FALSE,sep=",")
write.table(combined,file=file.path(INF,"work","pQTL-disease-GWAS-combined.csv"),row.names=FALSE,quote=FALSE,sep=",")
rxc
}
rxc <- gwas()3.2.3 Visualization
SF <- function(rxc, f="SF-pQTL-GWAS.png", ch=21, cw=21, h=13, w=18)
{
library(pheatmap)
col <- colorRampPalette(c("#4287f5","#ffffff","#e32222"))(3)
library(grid)
png(file.path(INF,f),res=300,width=w,height=h,units="in")
setHook("grid.newpage", function() pushViewport(viewport(x=1,y=1,width=0.9, height=0.9, name="vp", just=c("right","top"))), action="prepend")
colnames(rxc) <- gsub("^[0-9]*-","",colnames(rxc))
pheatmap(rxc, legend=FALSE, angle_col="270", border_color="black", color=col, cellheight=ch, cellwidth=cw, cluster_rows=TRUE, cluster_cols=FALSE, fontsize=8)
setHook("grid.newpage", NULL, "replace")
grid.text("Protein(s)-pQTL (gene)", y=0.03125, gp=gpar(fontsize=12))
grid.text("GWAS diseases", x=-0.0625, rot=90, gp=gpar(fontsize=12))
dev.off()
}
SF(rxc,f="SF-pQTL-GWAS.png",ch=8,cw=8,h=11,w=8.6)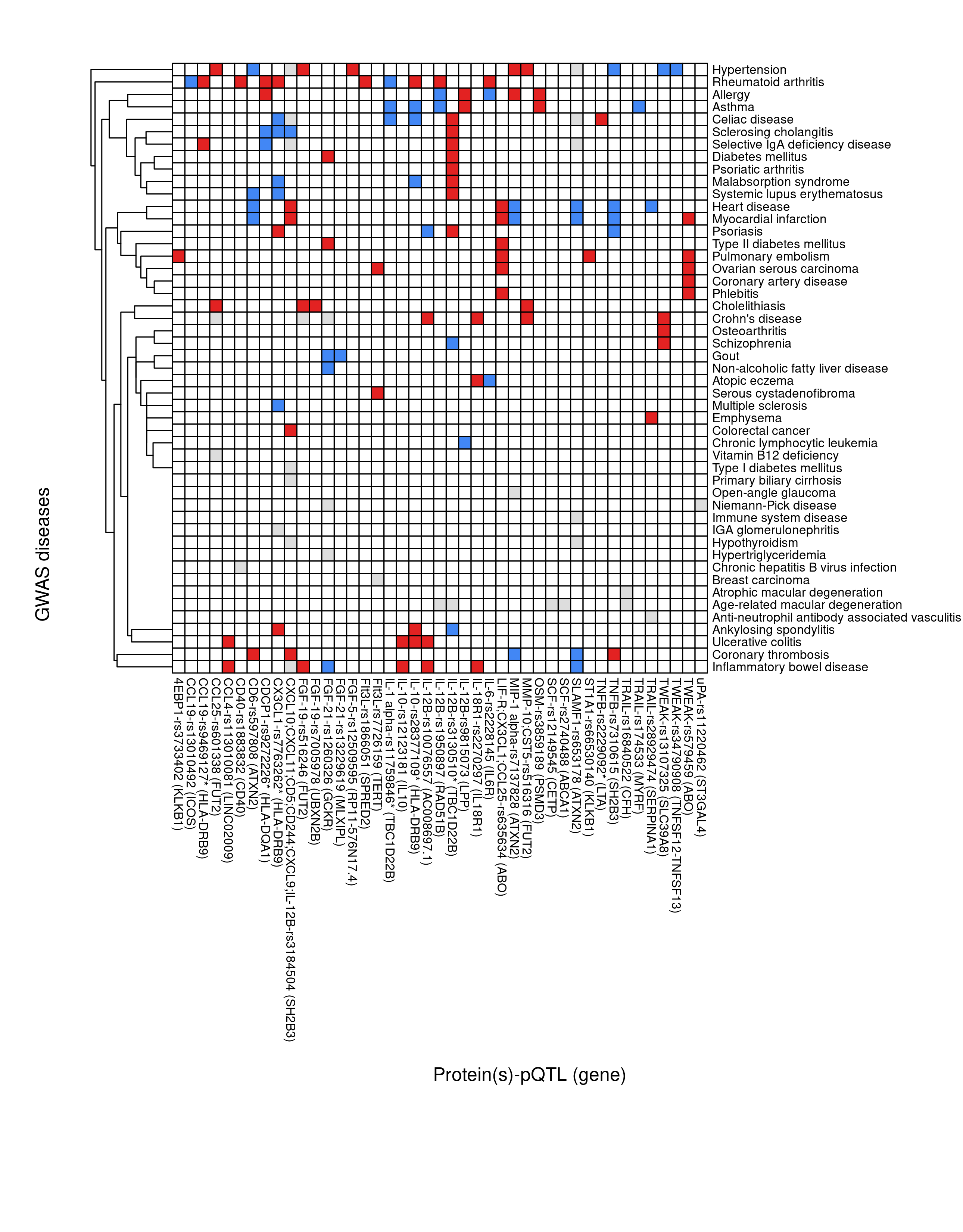
Figure 3.1: pQTL-disease overlap
4 Caprion analysis
This is from the Caprion project, https://jinghuazhao.github.io/Caprion/.
4.1 Colocalization
The coloc.R is modified slightly employing basename for local files.
liftRegion <- function(x, flanking = 1e6)
{
gr <- GenomicRanges::GRanges(seqnames = x$chr,
ranges = IRanges::IRanges(start = x$start - flanking, end = x$end + flanking))
seqlevelsStyle(gr) <- "UCSC"
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gr38 <- rtracklayer::liftOver(gr, chain)
lifted_chromosomes <- seqnames(gr38)
lifted_start <- min(start(gr38))
lifted_end <- max(end(gr38))
chr <- gsub("chr","",colnames(table(lifted_chromosomes)))
region <- paste0(chr, ":", lifted_start, "-", lifted_end)
invisible(list(chr = chr, start = lifted_start, end = lifted_end, region = region))
}
sumstats <- function(prot,chr,region37,chain)
{
cat("GWAS sumstats\n")
tbl <- file.path(analysis,"METAL_dr",paste0(prot,"_dr-1.tbl.gz"))
gwas_texts <- seqminer::tabix.read(tbl, tabixRange = region37)
gwas_stats <- read.table(text = gwas_texts, sep = "\t", header = FALSE) %>%
setNames(c("Chromosome","Position","ID","Allele1","Allele2","Freq1","FreqSE","MinFreq","MaxFreq",
"Effect","StdErr","logP","Direction","HetISq","HetChiSq","HetDf","logHetP","N"))
gwas_granges <- with(gwas_stats,GRanges(seqnames = paste0("chr",dplyr::if_else(Chromosome==23,"X",paste(Chromosome))),
ranges = IRanges(start = Position, end = Position),
id = ID,REF=Allele2,ALT=Allele1,AF=Freq1,ES=Effect,SE=StdErr,LP=-logP,SS=N))
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gwas_stats_hg38 <- rtracklayer::liftOver(gwas_granges, chain) %>%
unlist() %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames,
position = start, REF, ALT, AF, ES, SE, LP, SS) %>%
dplyr::mutate(id = paste(chromosome, position, sep = ":")) %>%
dplyr::mutate(MAF = pmin(AF, 1-AF)) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub("chr","",chromosome))
}
microarray <- function(gwas_stats_hg38,ensGene,region38)
{
cat("a. eQTL datasets\n")
microarray_df <- dplyr::filter(tabix_paths, quant_method == "microarray") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(microarray_df$ftp_path), microarray_df$qtl_id[1])
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.CEDAR")
column_names <- names(read.delim(hdr))
summary_list <- purrr::map(ftp_path_list, ~import_eQTLCatalogue(., region38,
selected_gene_id = ensGene, column_names))
purrr::map_df(summary_list[lapply(summary_list,nrow)!=0],
~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
rnaseq <- function(gwas_stats_hg38,ensGene,region38)
{
cat("b. Uniformly processed RNA-seq datasets\n")
rnaseq_df <- dplyr::filter(tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(rnaseq_df$ftp_path), rnaseq_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list, ~safe_import(., region38,
selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list[lapply(result_list,nrow)!=0], ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
purrr::map_df(result_list, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
gtex <- function(gwas_stats_hg38,ensGene,region38)
{
cat("c. GTEx_v8 imported eQTL datasets\n")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_gtex.tsv")
imported_tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>%
dplyr::mutate(ftp_path=file.path("~/rds/public_databases/GTEx/csv",basename(ftp_path)))
gtex_df <- dplyr::filter(imported_tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(gtex_df$ftp_path), gtex_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.GTEx")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
prot <- sentinels[["prot"]][r]
invisible(sapply(1:length(result_filtered), function(i) {
if (!is.null(result_filtered[[i]])) {
prot_name <- names(result_filtered)[i]
if (!is.null(prot_name)) {
f <- file.path(analysis, "coloc", "GTEx", "sumstats", paste0(prot, "-", prot_name, ".gz"))
gz <- gzfile(f, "w")
write.table(result_filtered[[i]], file = gz, col.names = TRUE, row.names = FALSE, quote = FALSE, sep = "\t")
close(gz)
} else {
warning(paste("Missing name for index", i))
}
}
}))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
ge <- function(gwas_stats_hg38,ensGene,region38)
{
cat("d. eQTL datasets\n")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_ge.tsv")
imported_tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>%
dplyr::mutate(ftp_path=file.path("~/rds/public_databases/eQTLCatalogue",basename(ftp_path)))
ftp_path_list <- setNames(as.list(imported_tabix_paths$ftp_path), imported_tabix_paths$unique_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
prot <- sentinels[["prot"]][r]
invisible(sapply(1:length(result_filtered), function(i) {
if (!is.null(result_filtered[[i]])) {
prot_name <- names(result_filtered)[i]
if (!is.null(prot_name)) {
f <- file.path(analysis, "coloc", "eQTLCatalogue", "sumstats", paste0(prot, "-", prot_name, ".gz"))
gz <- gzfile(f, "w")
write.table(result_filtered[[i]], file = gz, col.names = TRUE, row.names = FALSE, quote = FALSE, sep = "\t")
close(gz)
} else {
warning(paste("Missing name for index", i))
}
}
}))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "unique_id")
}
gtex_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37,chain)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
if (!exists("df_gtex")) return
saveRDS(df_gtex,file=paste0(out,".rds"))
p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram()
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggplot2::ggsave(plot = s, filename = paste0(out, ".assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggplot2::ggsave(plot = p, filename = paste0(out, ".hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
ge_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_ge <- ge(gwas_stats_hg38,ensGene,region38)
if (!exists("df_ge")) return
saveRDS(df_ge,file=paste0(out,".rds"))
p <- ggplot(df_ge, aes(x = PP.H4.abf)) + geom_histogram()
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, ".assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, ".hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
all_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_microarray <- microarray(gwas_stats_hg38,ensGene,region38)
df_rnaseq <- rnaseq(gwas_stats_hg38,ensGene,region38)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
df_ge <- ge(gwas_stats_hg38,ensGene,region38)
if (exists("df_microarray") & exits("df_rnaseq") & exists("df_gtex") & exists("df_ge"))
{
coloc_df = dplyr::bind_rows(df_microarray, df_rnaseq, df_gtex, df_ge)
saveRDS(coloc_df, file=paste0(out,"-all.rds"))
p <- ggplot(coloc_df, aes(x = PP.H4.abf)) + geom_histogram()
}
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, "-assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, "-hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
single_run <- function(r, batch="GTEx")
{
sentinel <- sentinels[r,]
prot <- sentinel[["prot"]]
chr <- sentinel[["geneChrom"]]
ensRegion37 <- with(sentinel,
{
start <- geneStart-M
if (start<0) start <- 0
end <- geneEnd+M
paste0(chr,":",start,"-",end)
})
ss <- subset(pQTLdata::caprion,Protein==paste0(sentinel[["prot"]],"_HUMAN"))
ensGene <- ss[["ensGenes"]]
x <- with(sentinel,list(chr=geneChrom,start=geneStart,end=geneEnd))
lr <- liftRegion(x)
ensRegion38 <- with(lr,paste0(chr,":",start-M,"-",end+M))
cat(chr,ensGene,ensRegion37,ensRegion38,"\n")
f <- file.path(analysis,"coloc",batch,with(sentinel,paste0(prot,"-",SNP)))
if (batch=="GTEx")
{
gtex_coloc(prot,chr,ensGene,chain,ensRegion37,ensRegion38,f)
} else {
ge_coloc(prot,chr,ensGene,chain,ensRegion37,ensRegion38,f)
}
}
collect <- function(batch="GTEx")
# to collect results when all single runs are done
{
df_coloc <- data.frame()
for(r in 1:nrow(sentinels))
{
prot <- sentinels[["prot"]][r]
snpid <- sentinels[["SNP"]][r]
rsid <- prot_rsid[["SNP"]][r]
f <- file.path(analysis,"coloc",batch,paste0(prot,"-",snpid,".rds"))
if (!file.exists(f)) next
cat(prot,"-",rsid,"\n")
rds <- readRDS(f)
if (nrow(rds)==0) next
df_coloc <- rbind(df_coloc,data.frame(prot=prot,rsid=rsid,snpid=snpid,rds))
}
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gr <- with(arrange(pQTLdata::caprion,Protein), {
GenomicRanges::GRanges(seqnames = chr,
ranges = IRanges::IRanges(start = start - M, end = end + M),
Protein=Protein,Gene=Gene)
})
seqlevelsStyle(gr) <- "UCSC"
gr38 <- rtracklayer::liftOver(gr,chain)
gr38_new <- lapply(gr38, function(gr) {
valid_start <- start(gr)[!is.na(start(gr))]
valid_end <- end(gr)[!is.na(end(gr))]
if (length(valid_start) > 0 && length(valid_end) > 0) {
min_start <- min(valid_start)
min_end <- min(valid_end)
new_gr <- GRanges(
seqnames = seqnames(gr)[1],
ranges = IRanges(start = min_start, end = min_end),
strand = strand(gr)[1],
Protein = ifelse(length(mcols(gr)$Protein) > 0, unique(mcols(gr)$Protein)[1], NA),
Gene = ifelse(length(mcols(gr)$Gene) > 0, unique(mcols(gr)$Gene)[1], NA)
)
return(new_gr)
} else {
return(NULL)
}
})
gr38_new <- gr38_new[!sapply(gr38_new, is.null)]
df38 <- do.call(rbind, lapply(gr38_new, function(gr) {
data.frame(
seqnames = as.character(seqnames(gr)),
start = start(gr),
end = end(gr),
strand = as.character(strand(gr)),
Protein = mcols(gr)$Protein,
Gene = mcols(gr)$Gene
)
})) %>%
mutate(range38=paste0(gsub("chr","",seqnames),":",start,"-",end)) %>%
select(Protein,Gene,range38)
caprion_upd <- pQTLdata::caprion %>%
mutate(prot=gsub("_HUMAN","",Protein),gene=Gene) %>%
left_join(df38)
df <- dplyr::rename(df_coloc,H0=PP.H0.abf,H1=PP.H1.abf,H2=PP.H2.abf,H3=PP.H3.abf,H4=PP.H4.abf) %>%
dplyr::left_join(caprion_upd[c("prot","gene","range38")])
if (batch=="GTEx") {
df_coloc <- within(df,{qtl_id <- gsub("GTEx_V8_","",qtl_id)})
write.table(subset(df,H4>=0.8),file=file.path(analysis,"coloc","GTEx.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
write.table(df,file=file.path(analysis,"coloc","GTEx-all.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
coloc <- merge(df_coloc,caprion_upd[c("prot","gene","range38")]) %>%
mutate(prot,
H0=round(H0,2),
H1=round(H1,2),
H2=round(H2,2),
H3=round(H3,2),
H4=round(H4,2)) %>%
setNames(c("Protein","Gene","RSid","SNPid","Tissue","nSNP","H0","H1","H2","H3","H4")) %>%
select(Protein,Gene,RSid,Tissue,nSNP,H0,H1,H2,H3,H4)
write.table(coloc,file=file.path(analysis,"coloc","GTEx-ST.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
} else {
write.table(subset(df,H4>=0.8),file=file.path(analysis,"coloc","eQTLCatalogue.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
write.table(df,file=file.path(analysis,"coloc","eQTLCatalogue-all.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
eQTLCatalogue <- left_join(df,caprion_upd[c("prot","gene","range38")]) %>%
mutate(prot,
H0=round(H0,2),
H1=round(H1,2),
H2=round(H2,2),
H3=round(H3,2),
H4=round(H4,2)) %>%
setNames(c("Protein","Gene","RSid","SNPid","Study","nSNP","H0","H1","H2","H3","H4")) %>%
select(Protein,Gene,RSid,Study,nSNP,H0,H1,H2,H3,H4)
write.table(eQTLCatalogue,file=file.path(analysis,"coloc","eQTLCatalogue-ST.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
}
}
loop_slowly <- function() for (r in 1:nrow(sentinels)) single_run(r)
# Environmental variables
pkgs <- c("dplyr", "gap", "ggplot2", "readr", "coloc", "GenomicRanges","pQTLtools","rtracklayer","seqminer")
invisible(suppressMessages(lapply(pkgs, require, character.only = TRUE)))
HOME <- Sys.getenv("HOME")
HPC_WORK <- Sys.getenv("HPC_WORK")
analysis <- Sys.getenv("analysis")
M <- 1e6
psum <- file.path(analysis,"coloc","sumstats")
if (!dir.exists(psum)) dir.create(psum)
gsum <- file.path(analysis,"coloc","GTEx","sumstats")
if (!dir.exists(gsum)) dir.create(gsum)
esum <- file.path(analysis,"coloc","eQTLCatalogue","sumstats")
if (!dir.exists(esum)) dir.create(esum)
sevens <- "
ENSG00000131142 - CCL25 19 8052318 8062660
ENSG00000125735 - TNFSF14 19 6661253 6670588
ENSG00000275302 - CCL4 17 36103827 36105621
ENSG00000274736 - CCL23 17 36013056 36017972
ENSG00000013725 - CD6 11 60971680 61020377
ENSG00000138675 - FGF5 4 80266639 80336680
ENSG00000277632 - CCL3 17 36088256 36090169
"
updates <- as.data.frame(scan(file=textConnection(sevens),what=list("","","",0,0,0))) %>%
setNames(c("ensGenes","dash","gene","chromosome","start38","end38"))
caprion <- left_join(pQTLdata::caprion,updates)
sentinels <- subset(read.csv(file.path(analysis,"work","caprion_dr.cis.vs.trans")),cis)
f <- file.path(analysis,"work","snpid_dr.lst")
prot_rsid <- select(sentinels,prot,SNP) %>%
dplyr::left_join(read.table(f,header=TRUE),by=c('SNP'='snpid')) %>%
transmute(prot,SNP=dplyr::if_else(is.na(rsid)|rsid==".",SNP,rsid))
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths.tsv")
tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>% dplyr::as_tibble()
r <- as.integer(Sys.getenv("r"))
single_run(r)
single_run(r,batch="eQTLCatalogue")
collect()
collect(batch="eQTLCatalogue")The single_run() is called for analysis with SLURM and collect() for summary with no need of SLURM.
4.2 LocusZoom.js
This actually involves liftOver back to GRCh37 done as follows for GTEx data.
awk 'NR>1{print $1,$2,$3,$4}' ${analysis}/coloc/GTEx.tsv | \
parallel -C ' ' '
export prot={1}
export rsid={2}
export snpid={3}
export tissue={4}
Rscript -e "
suppressMessages(library(dplyr))
suppressMessages(library(jsonlite))
suppressMessages(library(rtracklayer))
analysis <- Sys.getenv(\"analysis\")
hpc_work <- Sys.getenv(\"HPC_WORK\")
prot <- Sys.getenv(\"prot\")
rsid <- Sys.getenv(\"rsid\")
snpid <- Sys.getenv(\"snpid\")
tissue <- Sys.getenv(\"tissue\")
print(paste0(prot,\"-\",tissue))
f <- file.path(hpc_work,\"bin\",\"hg38ToHg19.over.chain\")
chain <- rtracklayer::import.chain(f)
liftOver <- function(gwas_stats)
{
gwas_granges <- with(gwas_stats,GRanges(seqnames = paste0(\"chr\",dplyr::if_else(chromosome==23,\"X\",paste(chromosome))),
ranges = IRanges(start = position, end = position),
chromosome = chromosome, position = position,
ref_allele=ref_allele,alt_allele=alt_allele,alt_allele_freq=alt_allele_freq,
log_pvalue=log_pvalue,beta=beta,se=se))
gwas_stats_hg37 <- rtracklayer::liftOver(gwas_granges, chain) %>%
unlist() %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames, position = start,
variant = paste0(chromosome,\":\",position,\"_\",ref_allele,\"/\",alt_allele),
ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
dplyr::mutate(id = paste(chromosome, position, sep = \":\")) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub(\"chr\",\"\",chromosome))
}
pGWAS_sumstats <- read.delim(file.path(analysis,\"coloc\",\"sumstats\",paste0(prot,\"-\",snpid,\".gz\"))) %>%
dplyr::mutate(REF=toupper(REF),ALT=toupper(ALT),variant=paste0(chromosome,\":\",position,\"_\",REF,\"/\",ALT)) %>%
dplyr::mutate(log_pvalue=LP,ref_allele=REF,alt_allele=ALT,alt_allele_freq=AF,beta=ES,se=SE) %>%
dplyr::select(chromosome,position,variant,ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
liftOver()
GTEx_sumstats <- read.delim(file.path(analysis,\"coloc\",\"GTEx\",\"sumstats\",paste0(prot,\"-\",tissue,\".gz\"))) %>%
dplyr::mutate(REF=toupper(ref),ALT=toupper(alt),variant=paste0(chromosome,\":\",position,\"_\",REF,\"/\",ALT)) %>%
dplyr::mutate(log_pvalue=-log10(pvalue),ref_allele=REF,alt_allele=ALT,alt_allele_freq=ac/an) %>%
dplyr::select(chromosome,position,variant,ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
liftOver()
j <- gzfile(file.path(analysis,\"json\",\"pqtleqtl\",paste0(prot,\"-\",tissue,\".json.gz\")))
combined_json <- jsonlite::toJSON(list(pqtl=pGWAS_sumstats,eqtl=GTEx_sumstats),auto_unbox=TRUE,pretty=TRUE)
write(combined_json,file=j)
close(j)
"
'Note that compressed json (.json.gz) is used.