This article provides details of pQTL/disease overlap and colocalization analysis.
1 pQTL/disease overlap
The ontology of traits/disease is available through Experimental Factor Ontology (EFO)1, which can be used to build lists of diseases and immune-mediated traits and filter search results from PhenoScanner2.
1.1 Diseases
library(ontologyIndex)
# http://www.ebi.ac.uk/efo/efo.obo
INF <- Sys.getenv("INF")
file <- file.path(INF,"ebi","efo-3.26.0","efo.obo")
get_relation_names(file)
efo <- get_ontology(file, extract_tags="everything")
id <- function(ontology)
{
length(ontology)
length(ontology$id)
inf <- grep(ontology$name,pattern="immune|inflammatory")
data.frame(id=ontology$id[inf],name=ontology$name[inf])
}
goidname <- id(go)
efoidname <- id(efo)
# all diseases
efo_diseases <- get_descendants(efo,"EFO:0000408")
diseases_name <- efo$name[efo_diseases]
diseases <- data.frame(efo_diseases,diseases_name)
write.table(diseases,file=file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=FALSE,row.names=FALSE,sep=",")
# immune system diseases (isd)
efo_0000540 <- get_descendants(efo,"EFO:0000540")
efo_0000540name <- efo$name[efo_0000540]
isd <- data.frame(efo_0000540,efo_0000540name)
library(ontologyPlot)
onto_plot(efo,efo_0000540)1.2 Lookup
options(width=200)
suppressMessages(library(dplyr))
suppressMessages(library(gap))
suppressMessages(library(pQTLtools))
inf1_prot <- vector()
for(i in 1:92) inf1_prot[inf1[i,"prot"]] <- mutate(inf1[i,],target.short=if_else(!is.na(alt_name),alt_name,target.short))[["target.short"]]
INF1_metal <- within(read.delim(file.path(find.package("pQTLtools"),"tests","INF1.METAL"),as.is=TRUE),{
hg19_coordinates=paste0("chr",Chromosome,":",Position)}) %>%
rename(INF1_rsid=rsid, Total=N) %>%
left_join(pQTLdata::inf1[c("prot","gene","target.short","alt_name")]) %>%
mutate(target.short=if_else(!is.na(alt_name),alt_name,target.short)) %>%
select(-alt_name)
INF1_aggr <- INF1_metal %>%
select(Chromosome,Position,target.short,gene,hg19_coordinates,
MarkerName,Allele1,Allele2,Freq1,Effect,StdErr,log.P.,cis.trans,INF1_rsid) %>%
group_by(Chromosome,Position,MarkerName,INF1_rsid,hg19_coordinates) %>%
summarise(nprots=n(),
prots=paste(target.short,collapse=";"),
Allele1=paste(toupper(Allele1),collapse=";"),
Allele2=paste(toupper(Allele2),collapse=";"),
EAF=paste(Freq1,collapse=";"),
Effects=paste(Effect,collapse=";"),
SEs=paste(StdErr,collapse=";"),
log10P=paste(log.P.,collapse=";"),
cistrans=paste(cis.trans,collapse=";")) %>%
data.frame()
rsid <- INF1_aggr[["INF1_rsid"]]
catalogue <- "GWAS"
proxies <- "EUR"
p <- 5e-8
r2 <- 0.8
build <- 37
INF <- Sys.getenv("INF")
efo_diseases <- read.table(file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=c("efo","disease"),as.is=TRUE,sep=",") %>%
mutate(efo=gsub(":", "_", efo))
r <- snpqueries(rsid, catalogue=catalogue, proxies=proxies, p=p, r2=r2, build=build)
lapply(r,dim)
snps_results <- with(r,right_join(snps,results))
ps <- subset(snps_results,select=-c(hg38_coordinates,ref_hg38_coordinates,pos_hg38,ref_pos_hg38,dprime))
aggr <- subset(within(INF1_aggr,{HLA <- as.numeric(Chromosome==6 & Position >= 25392021 & Position <= 33392022)}),
select=-c(Chromosome,Position,INF1_rsid))
short <- merge(aggr,ps,by="hg19_coordinates")
gwas <- function()
{
short <- merge(aggr,ps,by="hg19_coordinates") %>%
filter(efo %in% pull(efo_diseases,efo)) %>%
left_join(efo_diseases)
v <- c("prots","hgnc","MarkerName","cistrans","Effects","Allele1","Allele2","rsid","a1","a2","efo",
"ref_rsid","ref_a1","ref_a2","proxy","r2",
"HLA","beta","se","p","disease","n_cases","n_controls","unit","ancestry","pmid","study")
mat <- within(short[v],
{
flag <- (HLA==1)
prefix <- paste0(prots,"-",rsid)
prefix[flag] <- paste0(prefix[flag],"*")
rsidProts <- paste0(prefix," (",hgnc,")")
efoTraits <- gsub("\\b(^[a-z])","\\U\\1",disease,perl=TRUE)
qtl_direction <- sign(as.numeric(beta))
})
combined <- group_by(mat,efoTraits,rsidProts,desc(n_cases)) %>%
summarize(direction=paste(qtl_direction,collapse=";"),
betas=paste(beta,collapse=";"),
units=paste(unit,collapse=";"),
studies=paste(study,collapse=";"),
diseases=paste(disease,collapse=";"),
cases=paste(n_cases,collapse=";")
) %>%
data.frame()
rxc <- with(combined,table(efoTraits,rsidProts))
for(cn in colnames(rxc)) for(rn in rownames(rxc)) {
cnrn <- subset(combined,efoTraits==rn & rsidProts==cn)
if(nrow(cnrn)==0) next
rxc[rn,cn] <- as.numeric(unlist(strsplit(cnrn[["direction"]],";"))[1])
}
write.table(mat,file=file.path(INF,"work","pQTL-disease-GWAS.csv"),row.names=FALSE,quote=FALSE,sep=",")
write.table(combined,file=file.path(INF,"work","pQTL-disease-GWAS-combined.csv"),row.names=FALSE,quote=FALSE,sep=",")
rxc
}
rxc <- gwas()1.3 Visualization
SF <- function(rxc, f="SF-pQTL-GWAS.png", ch=21, cw=21, h=13, w=18)
{
library(pheatmap)
col <- colorRampPalette(c("#4287f5","#ffffff","#e32222"))(3)
library(grid)
png(file.path(INF,f),res=300,width=w,height=h,units="in")
setHook("grid.newpage", function() pushViewport(viewport(x=1,y=1,width=0.9, height=0.9, name="vp", just=c("right","top"))), action="prepend")
colnames(rxc) <- gsub("^[0-9]*-","",colnames(rxc))
pheatmap(rxc, legend=FALSE, angle_col="270", border_color="black", color=col, cellheight=ch, cellwidth=cw, cluster_rows=TRUE, cluster_cols=FALSE, fontsize=8)
setHook("grid.newpage", NULL, "replace")
grid.text("Protein(s)-pQTL (gene)", y=0.03125, gp=gpar(fontsize=12))
grid.text("GWAS diseases", x=-0.0625, rot=90, gp=gpar(fontsize=12))
dev.off()
}
SF(rxc,f="SF-pQTL-GWAS.png",ch=8,cw=8,h=11,w=8.6)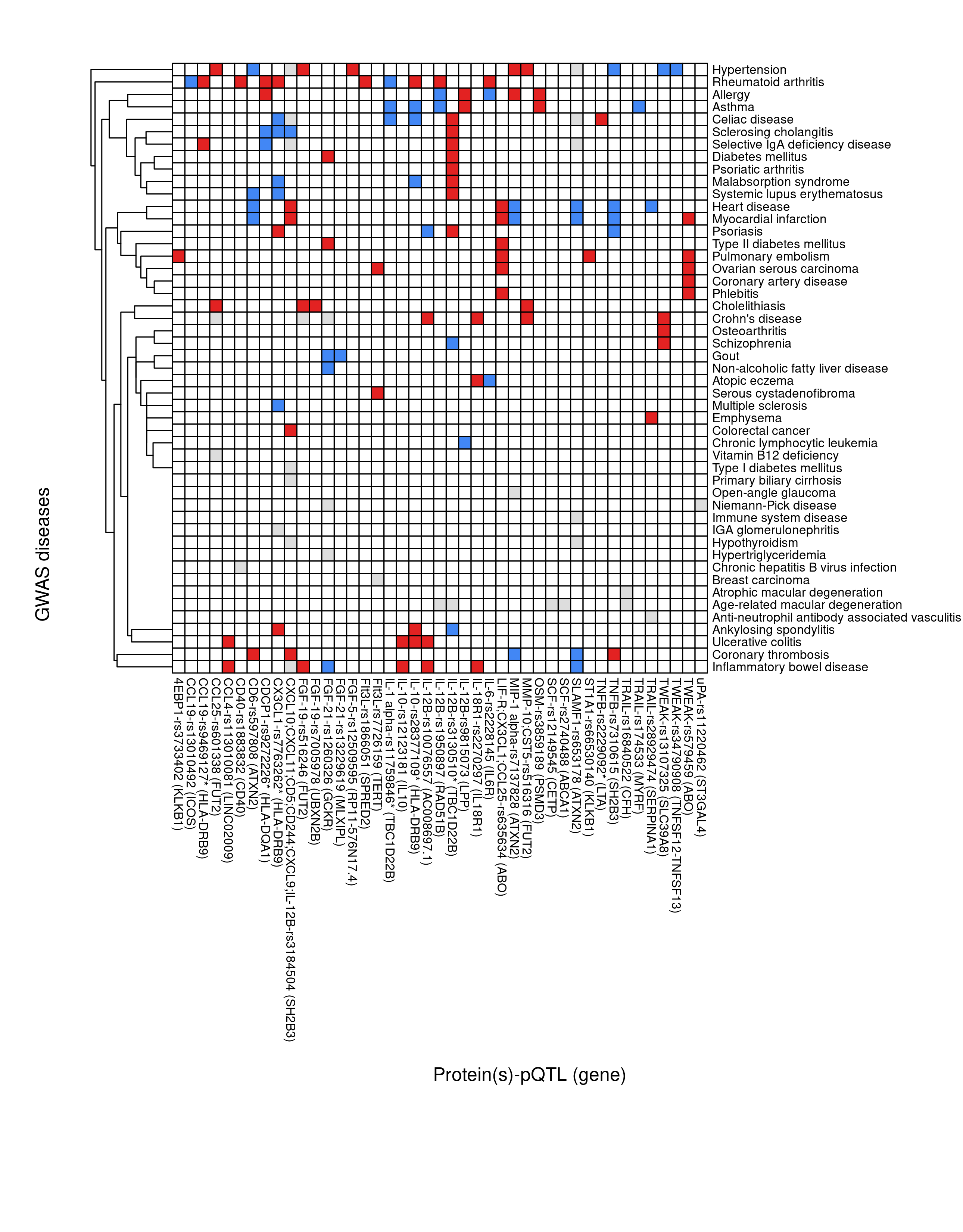
Figure 1.1: pQTL-disease overlap
2 Colocalization
This is the actual script for cis-pQTL colocalization analysis on GTEx v8 for SCALLOP-INF.
2.1 Data
The data were GWAS summary statistics in GRCh37 and VCF format, converted by gwasvcf. The GTEx association statistics were in GRCh38 and downloaded from the eQTL Catalogue and stored locally. Data on microarray and RNA-Seq remain on the eQTL Catalogue website.
2.2 coloc.R
It contains minor modification to the documentation example,
liftRegion <- function(x,chain,flanking=1e6)
{
require(GenomicRanges)
gr <- with(x,GenomicRanges::GRanges(seqnames=chr,IRanges::IRanges(start,end))+flanking)
seqlevelsStyle(gr) <- "UCSC"
gr38 <- rtracklayer::liftOver(gr, chain)
chr <- gsub("chr","",colnames(table(seqnames(gr38))))
start <- min(unlist(start(gr38)))
end <- max(unlist(end(gr38)))
invisible(list(chr=chr[1],start=start,end=end,region=paste0(chr,":",start,"-",end)))
}
sumstats <- function(prot,chr,region37)
{
cat("GWAS sumstats\n")
vcf <- file.path(INF,"METAL/gwas2vcf",paste0(prot,".vcf.gz"))
gwas_stats <- gwasvcf::query_gwas(vcf, chrompos = region37) %>%
gwasvcf::vcf_to_granges() %>%
keepSeqlevels(chr) %>%
renameSeqlevels(paste0("chr",chr))
gwas_stats_hg38 <- rtracklayer::liftOver(gwas_stats, chain) %>%
unlist() %>%
# renameSeqlevels(chr) %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames,
position = start, REF, ALT, AF, ES, SE, LP, SS) %>%
dplyr::mutate(id = paste(chromosome, position, sep = ":")) %>%
dplyr::mutate(MAF = pmin(AF, 1-AF)) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub("chr","",chromosome))
}
microarray <- function(gwas_stats_hg38,ensGene,region38)
{
cat("a. eQTL datasets\n")
microarray_df <- dplyr::filter(tabix_paths, quant_method == "microarray") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(microarray_df$ftp_path), microarray_df$qtl_id[1])
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.CEDAR")
column_names <- names(read.delim(hdr))
summary_list <- purrr::map(ftp_path_list, ~import_eQTLCatalogue(., region38,
selected_gene_id = ensGene, column_names))
purrr::map_df(summary_list[lapply(summary_list,nrow)!=0],
~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
rnaseq <- function(gwas_stats_hg38,ensGene,region38)
{
cat("b. Uniformly processed RNA-seq datasets\n")
rnaseq_df <- dplyr::filter(tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(rnaseq_df$ftp_path), rnaseq_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list, ~safe_import(., region38,
selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list[lapply(result_list,nrow)!=0], ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
purrr::map_df(result_list, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
gtex <- function(gwas_stats_hg38,ensGene,region38)
{
cat("c. GTEx_v8 imported eQTL datasets\n")
gtex_df <- dplyr::filter(imported_tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(gtex_df$ftp_path), gtex_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.GTEx")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
coloc <- function(prot,chr,ensGene,chain,region37,region38,out,run_all=FALSE)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
if (exists("df_gtex"))
{
saveRDS(df_gtex,file=paste0(out,".RDS"))
dplyr::arrange(df_gtex, -PP.H4.abf)
p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram()
}
if (run_all)
{
df_microarray <- microarray(gwas_stats_hg38,ensGene,region38)
df_rnaseq <- rnaseq(gwas_stats_hg38,ensGene,region38)
if (exists("df_microarray") & exits("df_rnaseq") & exists("df_gtex"))
{
coloc_df = dplyr::bind_rows(df_microarray, df_rnaseq, df_gtex)
saveRDS(coloc_df, file=paste0(out,".RDS"))
dplyr::arrange(coloc_df, -PP.H4.abf)
p <- ggplot(coloc_df, aes(x = PP.H4.abf)) + geom_histogram()
}
}
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, "-assoc.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, "-hist.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
single_run <- function(r)
{
sentinel <- sentinels[r,]
chr <- with(sentinel,Chr)
ss <- subset(inf1,prot==sentinel[["prot"]])
ensRegion37 <- with(ss,
{
start <- start-M
if (start<0) start <- 0
end <- end+M
paste0(chr,":",start,"-",end)
})
ensGene <- ss[["ensembl_gene_id"]]
ensRegion38 <- with(liftRegion(ss,chain),region)
f <- file.path(INF,"coloc",with(sentinel,paste0(prot,"-",SNP)))
cat(chr,ensGene,ensRegion37,ensRegion38,"\n")
coloc(sentinel[["prot"]],chr,ensGene,chain,ensRegion37,ensRegion38,f)
}
# slow with the following loop:
loop <- function() for (r in 1:nrow(sentinels)) single_run(r)
library(pQTLtools)
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
pkgs <- c("dplyr", "ggplot2", "readr", "coloc", "GenomicRanges","seqminer")
invisible(lapply(pkgs, require, character.only = TRUE))
HPC_WORK <- Sys.getenv("HPC_WORK")
gwasvcf::set_bcftools(file.path(HPC_WORK,"bin","bcftools"))
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths.tsv")
tabix_paths <- read.delim(f, stringsAsFactors = FALSE) %>% dplyr::as_tibble()
HOME <- Sys.getenv("HOME")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_gtex.tsv")
imported_tabix_paths <- within(read.delim(fp, stringsAsFactors = FALSE) %>% dplyr::as_tibble(),
{
f <- lapply(strsplit(ftp_path,"/csv/|/ge/"),"[",3)
ftp_path <- paste0("~/rds/public_databases/GTEx/csv"),f)
})
options(width=200)
library(dplyr)
INF <- Sys.getenv("INF")
M <- 1e6
sentinels <- subset(read.csv(file.path(INF,"work","INF1.merge.cis.vs.trans")),cis)
cvt_rsid <- file.path(INF,"work","INF1.merge.cis.vs.trans-rsid")
prot_rsid <- subset(read.delim(cvt_rsid,sep=" "),cis,select=c(prot,SNP))
# Faster with parallel Bash runs.
r <- as.integer(Sys.getenv("r"))
single_run(r)where options for protein GWAS, microarray, RNA-Seq are available with respect to variant-flanking or gene regions. When no results are generated,
there would have problem with dplyr::arrange(df_gtex, -PP.H4.abf);p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram().
2.3 Collection of results
When these are furnished we keep results (i.e., PP4>=0.8) as follows,
collect <- function()
{
df_coloc <- data.frame()
for(r in 1:nrow(sentinels))
{
prot <- sentinels[["prot"]][r]
snpid <- sentinels[["SNP"]][r]
rsid <- prot_rsid[["SNP"]][r]
f <- file.path(INF,"coloc",paste0(prot,"-",snpid,".RDS"))
if (!file.exists(f)) next
cat(prot,"-",rsid,"\n")
rds <- readRDS(f)
if (nrow(rds)==0) next
df_coloc <- rbind(df_coloc,data.frame(prot=prot,rsid=rsid,snpid=snpid,rds))
}
df_coloc <- within(df_coloc,{qtl_id <- gsub("GTEx_V8_","",qtl_id)}) %>%
rename(H0=PP.H0.abf,H1=PP.H1.abf,H2=PP.H2.abf,H3=PP.H3.abf,H4=PP.H4.abf)
write.table(subset(df_coloc,H4>=0.8),
file=file.path(INF,"coloc","GTEx.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
}
collect()2.4 The driver program
It is in Bash.
#!/usr/bin/bash
for r in {1..59}
do
export r=${r}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
done2.5 Parallel computing
To speed up the analysis, we resort to SLURM,
#!/usr/bin/bash
#SBATCH --job-name=_coloc
#SBATCH --account CARDIO-SL0-CPU
#SBATCH --partition cardio
#SBATCH --qos=cardio
#SBATCH --array=1-59
#SBATCH --mem=28800
#SBATCH --time=5-00:00:00
#SBATCH --error=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.err
#SBATCH --output=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.out
#SBATCH --export ALL
export trait=$(awk 'NR==ENVIRON["SLURM_ARRAY_TASK_ID"] {print $1}' ${INF}/work/inf1.tmp)
function gtex()
{
export r=${SLURM_ARRAY_TASK_ID}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
}
gtex