This article provides details of pQTL/disease overlap and colocalization analysis.
1 SCALLOP analysis
1.1 pQTL/disease overlap
The ontology of traits/disease is available through Experimental Factor Ontology (EFO)1, which can be used to build lists of diseases and immune-mediated traits and filter search results from PhenoScanner2.
1.1.1 Diseases
library(ontologyIndex)
# http://www.ebi.ac.uk/efo/efo.obo
INF <- Sys.getenv("INF")
file <- file.path(INF,"ebi","efo-3.26.0","efo.obo")
get_relation_names(file)
efo <- get_ontology(file, extract_tags="everything")
id <- function(ontology)
{
length(ontology)
length(ontology$id)
inf <- grep(ontology$name,pattern="immune|inflammatory")
data.frame(id=ontology$id[inf],name=ontology$name[inf])
}
goidname <- id(go)
efoidname <- id(efo)
# all diseases
efo_diseases <- get_descendants(efo,"EFO:0000408")
diseases_name <- efo$name[efo_diseases]
diseases <- data.frame(efo_diseases,diseases_name)
write.table(diseases,file=file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=FALSE,row.names=FALSE,sep=",")
# immune system diseases (isd)
efo_0000540 <- get_descendants(efo,"EFO:0000540")
efo_0000540name <- efo$name[efo_0000540]
isd <- data.frame(efo_0000540,efo_0000540name)
library(ontologyPlot)
onto_plot(efo,efo_0000540)1.1.2 Lookup
options(width=200)
suppressMessages(library(dplyr))
suppressMessages(library(gap))
suppressMessages(library(pQTLtools))
inf1_prot <- vector()
for(i in 1:92) inf1_prot[inf1[i,"prot"]] <- mutate(inf1[i,],target.short=if_else(!is.na(alt_name),alt_name,target.short))[["target.short"]]
INF1_metal <- within(read.delim(file.path(find.package("pQTLtools"),"tests","INF1.METAL"),as.is=TRUE),{
hg19_coordinates=paste0("chr",Chromosome,":",Position)}) %>%
rename(INF1_rsid=rsid, Total=N) %>%
left_join(pQTLdata::inf1[c("prot","gene","target.short","alt_name")]) %>%
mutate(target.short=if_else(!is.na(alt_name),alt_name,target.short)) %>%
select(-alt_name)
INF1_aggr <- INF1_metal %>%
select(Chromosome,Position,target.short,gene,hg19_coordinates,
MarkerName,Allele1,Allele2,Freq1,Effect,StdErr,log.P.,cis.trans,INF1_rsid) %>%
group_by(Chromosome,Position,MarkerName,INF1_rsid,hg19_coordinates) %>%
summarise(nprots=n(),
prots=paste(target.short,collapse=";"),
Allele1=paste(toupper(Allele1),collapse=";"),
Allele2=paste(toupper(Allele2),collapse=";"),
EAF=paste(Freq1,collapse=";"),
Effects=paste(Effect,collapse=";"),
SEs=paste(StdErr,collapse=";"),
log10P=paste(log.P.,collapse=";"),
cistrans=paste(cis.trans,collapse=";")) %>%
data.frame()
rsid <- INF1_aggr[["INF1_rsid"]]
catalogue <- "GWAS"
proxies <- "EUR"
p <- 5e-8
r2 <- 0.8
build <- 37
INF <- Sys.getenv("INF")
efo_diseases <- read.table(file.path(INF,"ebi","efo-3.26.0","efo_diseases.csv"),col.names=c("efo","disease"),as.is=TRUE,sep=",") %>%
mutate(efo=gsub(":", "_", efo))
r <- snpqueries(rsid, catalogue=catalogue, proxies=proxies, p=p, r2=r2, build=build)
lapply(r,dim)
snps_results <- with(r,right_join(snps,results))
ps <- subset(snps_results,select=-c(hg38_coordinates,ref_hg38_coordinates,pos_hg38,ref_pos_hg38,dprime))
aggr <- subset(within(INF1_aggr,{HLA <- as.numeric(Chromosome==6 & Position >= 25392021 & Position <= 33392022)}),
select=-c(Chromosome,Position,INF1_rsid))
short <- merge(aggr,ps,by="hg19_coordinates")
gwas <- function()
{
short <- merge(aggr,ps,by="hg19_coordinates") %>%
filter(efo %in% pull(efo_diseases,efo)) %>%
left_join(efo_diseases)
v <- c("prots","hgnc","MarkerName","cistrans","Effects","Allele1","Allele2","rsid","a1","a2","efo",
"ref_rsid","ref_a1","ref_a2","proxy","r2",
"HLA","beta","se","p","disease","n_cases","n_controls","unit","ancestry","pmid","study")
mat <- within(short[v],
{
flag <- (HLA==1)
prefix <- paste0(prots,"-",rsid)
prefix[flag] <- paste0(prefix[flag],"*")
rsidProts <- paste0(prefix," (",hgnc,")")
efoTraits <- gsub("\\b(^[a-z])","\\U\\1",disease,perl=TRUE)
qtl_direction <- sign(as.numeric(beta))
})
combined <- group_by(mat,efoTraits,rsidProts,desc(n_cases)) %>%
summarize(direction=paste(qtl_direction,collapse=";"),
betas=paste(beta,collapse=";"),
units=paste(unit,collapse=";"),
studies=paste(study,collapse=";"),
diseases=paste(disease,collapse=";"),
cases=paste(n_cases,collapse=";")
) %>%
data.frame()
rxc <- with(combined,table(efoTraits,rsidProts))
for(cn in colnames(rxc)) for(rn in rownames(rxc)) {
cnrn <- subset(combined,efoTraits==rn & rsidProts==cn)
if(nrow(cnrn)==0) next
rxc[rn,cn] <- as.numeric(unlist(strsplit(cnrn[["direction"]],";"))[1])
}
write.table(mat,file=file.path(INF,"work","pQTL-disease-GWAS.csv"),row.names=FALSE,quote=FALSE,sep=",")
write.table(combined,file=file.path(INF,"work","pQTL-disease-GWAS-combined.csv"),row.names=FALSE,quote=FALSE,sep=",")
rxc
}
rxc <- gwas()1.1.3 Visualization
SF <- function(rxc, f="SF-pQTL-GWAS.png", ch=21, cw=21, h=13, w=18)
{
library(pheatmap)
col <- colorRampPalette(c("#4287f5","#ffffff","#e32222"))(3)
library(grid)
png(file.path(INF,f),res=300,width=w,height=h,units="in")
setHook("grid.newpage", function() pushViewport(viewport(x=1,y=1,width=0.9, height=0.9, name="vp", just=c("right","top"))), action="prepend")
colnames(rxc) <- gsub("^[0-9]*-","",colnames(rxc))
pheatmap(rxc, legend=FALSE, angle_col="270", border_color="black", color=col, cellheight=ch, cellwidth=cw, cluster_rows=TRUE, cluster_cols=FALSE, fontsize=8)
setHook("grid.newpage", NULL, "replace")
grid.text("Protein(s)-pQTL (gene)", y=0.03125, gp=gpar(fontsize=12))
grid.text("GWAS diseases", x=-0.0625, rot=90, gp=gpar(fontsize=12))
dev.off()
}
SF(rxc,f="SF-pQTL-GWAS.png",ch=8,cw=8,h=11,w=8.6)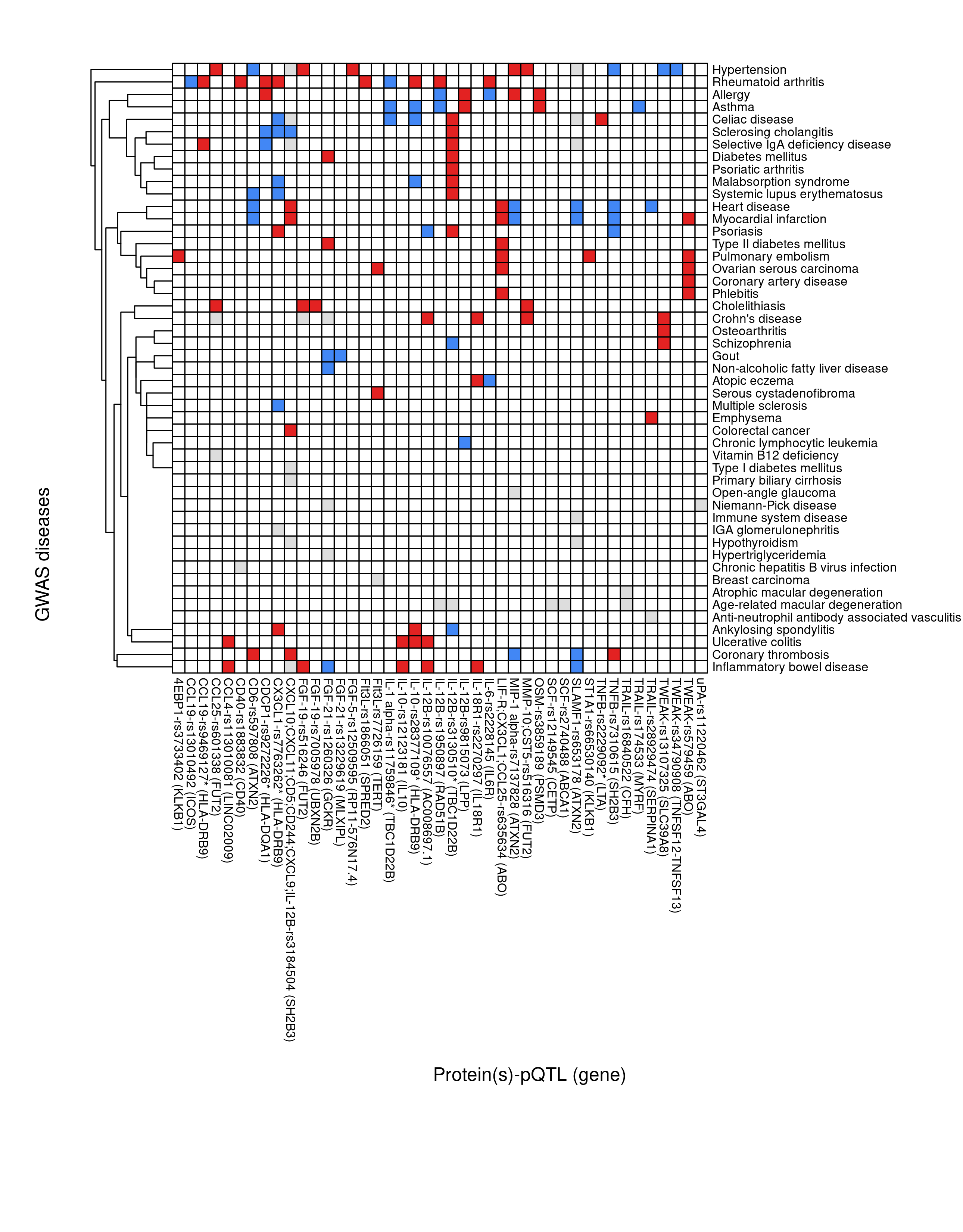
Figure 1.1: pQTL-disease overlap
1.2 Colocalization
This is the actual script for cis-pQTL colocalization analysis on GTEx v8 for SCALLOP-INF.
1.2.1 Data
The data were GWAS summary statistics in GRCh37 and VCF format, converted by gwasvcf. The GTEx association statistics were in GRCh38 and downloaded from the eQTL Catalogue and stored locally. Data on microarray and RNA-Seq remain on the eQTL Catalogue website.
1.2.2 coloc.R
It contains minor modification to the documentation example,
liftRegion <- function(x,chain,flanking=1e6)
{
require(GenomicRanges)
gr <- with(x,GenomicRanges::GRanges(seqnames=chr,IRanges::IRanges(start,end))+flanking)
seqlevelsStyle(gr) <- "UCSC"
gr38 <- rtracklayer::liftOver(gr, chain)
chr <- gsub("chr","",colnames(table(seqnames(gr38))))
start <- min(unlist(start(gr38)))
end <- max(unlist(end(gr38)))
invisible(list(chr=chr[1],start=start,end=end,region=paste0(chr,":",start,"-",end)))
}
sumstats <- function(prot,chr,region37)
{
cat("GWAS sumstats\n")
vcf <- file.path(INF,"METAL/gwas2vcf",paste0(prot,".vcf.gz"))
gwas_stats <- gwasvcf::query_gwas(vcf, chrompos = region37) %>%
gwasvcf::vcf_to_granges() %>%
keepSeqlevels(chr) %>%
renameSeqlevels(paste0("chr",chr))
gwas_stats_hg38 <- rtracklayer::liftOver(gwas_stats, chain) %>%
unlist() %>%
# renameSeqlevels(chr) %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames,
position = start, REF, ALT, AF, ES, SE, LP, SS) %>%
dplyr::mutate(id = paste(chromosome, position, sep = ":")) %>%
dplyr::mutate(MAF = pmin(AF, 1-AF)) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub("chr","",chromosome))
}
microarray <- function(gwas_stats_hg38,ensGene,region38)
{
cat("a. eQTL datasets\n")
microarray_df <- dplyr::filter(tabix_paths, quant_method == "microarray") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(microarray_df$ftp_path), microarray_df$qtl_id[1])
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.CEDAR")
column_names <- names(read.delim(hdr))
summary_list <- purrr::map(ftp_path_list, ~import_eQTLCatalogue(., region38,
selected_gene_id = ensGene, column_names))
purrr::map_df(summary_list[lapply(summary_list,nrow)!=0],
~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
rnaseq <- function(gwas_stats_hg38,ensGene,region38)
{
cat("b. Uniformly processed RNA-seq datasets\n")
rnaseq_df <- dplyr::filter(tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(rnaseq_df$ftp_path), rnaseq_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list, ~safe_import(., region38,
selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list[lapply(result_list,nrow)!=0], ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
purrr::map_df(result_list, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
gtex <- function(gwas_stats_hg38,ensGene,region38)
{
cat("c. GTEx_v8 imported eQTL datasets\n")
gtex_df <- dplyr::filter(imported_tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(gtex_df$ftp_path), gtex_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.GTEx")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
coloc <- function(prot,chr,ensGene,chain,region37,region38,out,run_all=FALSE)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
if (exists("df_gtex"))
{
saveRDS(df_gtex,file=paste0(out,".RDS"))
dplyr::arrange(df_gtex, -PP.H4.abf)
p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram()
}
if (run_all)
{
df_microarray <- microarray(gwas_stats_hg38,ensGene,region38)
df_rnaseq <- rnaseq(gwas_stats_hg38,ensGene,region38)
if (exists("df_microarray") & exits("df_rnaseq") & exists("df_gtex"))
{
coloc_df = dplyr::bind_rows(df_microarray, df_rnaseq, df_gtex)
saveRDS(coloc_df, file=paste0(out,".RDS"))
dplyr::arrange(coloc_df, -PP.H4.abf)
p <- ggplot(coloc_df, aes(x = PP.H4.abf)) + geom_histogram()
}
}
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, "-assoc.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, "-hist.pdf"), path = "", device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
single_run <- function(r)
{
sentinel <- sentinels[r,]
chr <- with(sentinel,Chr)
ss <- subset(inf1,prot==sentinel[["prot"]])
ensRegion37 <- with(ss,
{
start <- start-M
if (start<0) start <- 0
end <- end+M
paste0(chr,":",start,"-",end)
})
ensGene <- ss[["ensembl_gene_id"]]
ensRegion38 <- with(liftRegion(ss,chain),region)
f <- file.path(INF,"coloc",with(sentinel,paste0(prot,"-",SNP)))
cat(chr,ensGene,ensRegion37,ensRegion38,"\n")
coloc(sentinel[["prot"]],chr,ensGene,chain,ensRegion37,ensRegion38,f)
}
# slow with the following loop:
loop <- function() for (r in 1:nrow(sentinels)) single_run(r)
library(pQTLtools)
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
pkgs <- c("dplyr", "ggplot2", "readr", "coloc", "GenomicRanges","seqminer")
invisible(lapply(pkgs, require, character.only = TRUE))
HPC_WORK <- Sys.getenv("HPC_WORK")
gwasvcf::set_bcftools(file.path(HPC_WORK,"bin","bcftools"))
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths.tsv")
tabix_paths <- read.delim(f, stringsAsFactors = FALSE) %>% dplyr::as_tibble()
HOME <- Sys.getenv("HOME")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_gtex.tsv")
imported_tabix_paths <- within(read.delim(fp, stringsAsFactors = FALSE) %>% dplyr::as_tibble(),
{
f <- lapply(strsplit(ftp_path,"/csv/|/ge/"),"[",3)
ftp_path <- paste0("~/rds/public_databases/GTEx/csv"),f)
})
options(width=200)
library(dplyr)
INF <- Sys.getenv("INF")
M <- 1e6
sentinels <- subset(read.csv(file.path(INF,"work","INF1.merge.cis.vs.trans")),cis)
cvt_rsid <- file.path(INF,"work","INF1.merge.cis.vs.trans-rsid")
prot_rsid <- subset(read.delim(cvt_rsid,sep=" "),cis,select=c(prot,SNP))
# Faster with parallel Bash runs.
r <- as.integer(Sys.getenv("r"))
single_run(r)where options for protein GWAS, microarray, RNA-Seq are available with respect to variant-flanking or gene regions. When no results are generated,
there would have problem with dplyr::arrange(df_gtex, -PP.H4.abf);p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram().
1.2.3 Collection of results
When these are furnished we keep results (i.e., PP4>=0.8) as follows,
collect <- function()
{
df_coloc <- data.frame()
for(r in 1:nrow(sentinels))
{
prot <- sentinels[["prot"]][r]
snpid <- sentinels[["SNP"]][r]
rsid <- prot_rsid[["SNP"]][r]
f <- file.path(INF,"coloc",paste0(prot,"-",snpid,".RDS"))
if (!file.exists(f)) next
cat(prot,"-",rsid,"\n")
rds <- readRDS(f)
if (nrow(rds)==0) next
df_coloc <- rbind(df_coloc,data.frame(prot=prot,rsid=rsid,snpid=snpid,rds))
}
df_coloc <- within(df_coloc,{qtl_id <- gsub("GTEx_V8_","",qtl_id)}) %>%
rename(H0=PP.H0.abf,H1=PP.H1.abf,H2=PP.H2.abf,H3=PP.H3.abf,H4=PP.H4.abf)
write.table(subset(df_coloc,H4>=0.8),
file=file.path(INF,"coloc","GTEx.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
}
collect()1.3 The driver program
It is in Bash.
#!/usr/bin/bash
for r in {1..59}
do
export r=${r}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
done1.3.1 Parallel computing
To speed up the analysis, we resort to SLURM,
#!/usr/bin/bash
#SBATCH --job-name=_coloc
#SBATCH --account CARDIO-SL0-CPU
#SBATCH --partition cardio
#SBATCH --qos=cardio
#SBATCH --array=1-59
#SBATCH --mem=28800
#SBATCH --time=5-00:00:00
#SBATCH --error=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.err
#SBATCH --output=/rds/user/jhz22/hpc-work/work/_coloc_%A_%a.out
#SBATCH --export ALL
export trait=$(awk 'NR==ENVIRON["SLURM_ARRAY_TASK_ID"] {print $1}' ${INF}/work/inf1.tmp)
function gtex()
{
export r=${SLURM_ARRAY_TASK_ID}
export cvt=${INF}/work/INF1.merge.cis.vs.trans
read prot MarkerName < \
<(awk -vFS="," '$14=="cis"' ${cvt} | \
awk -vFS="," -vr=${r} 'NR==r{print $2,$5}')
echo ${r} - ${prot} - ${MarkerName}
export prot=${prot}
export MarkerName=${MarkerName}
if [ ! -f ${INF}/coloc/${prot}-${MarkerName}.pdf ] || \
[ ! -f ${INF}/coloc/${prot}-${MarkerName}.RDS ]; then
cd ${INF}/coloc
R --no-save < ${INF}/rsid/coloc.R 2>&1 | \
tee ${prot}-${MarkerName}.log
cd -
fi
}
gtex2 Caprion analysis
This is from the Caprion project, https://jinghuazhao.github.io/Caprion/.
2.1 Colocalization
The coloc.R is modified slightly employing basename for local files.
liftRegion <- function(x, flanking = 1e6)
{
gr <- GenomicRanges::GRanges(seqnames = x$chr,
ranges = IRanges::IRanges(start = x$start - flanking, end = x$end + flanking))
seqlevelsStyle(gr) <- "UCSC"
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gr38 <- rtracklayer::liftOver(gr, chain)
lifted_chromosomes <- seqnames(gr38)
lifted_start <- min(start(gr38))
lifted_end <- max(end(gr38))
chr <- gsub("chr","",colnames(table(lifted_chromosomes)))
region <- paste0(chr, ":", lifted_start, "-", lifted_end)
invisible(list(chr = chr, start = lifted_start, end = lifted_end, region = region))
}
sumstats <- function(prot,chr,region37,chain)
{
cat("GWAS sumstats\n")
tbl <- file.path(analysis,"METAL_dr",paste0(prot,"_dr-1.tbl.gz"))
gwas_texts <- seqminer::tabix.read(tbl, tabixRange = region37)
gwas_stats <- read.table(text = gwas_texts, sep = "\t", header = FALSE) %>%
setNames(c("Chromosome","Position","ID","Allele1","Allele2","Freq1","FreqSE","MinFreq","MaxFreq",
"Effect","StdErr","logP","Direction","HetISq","HetChiSq","HetDf","logHetP","N"))
gwas_granges <- with(gwas_stats,GRanges(seqnames = paste0("chr",dplyr::if_else(Chromosome==23,"X",paste(Chromosome))),
ranges = IRanges(start = Position, end = Position),
id = ID,REF=Allele2,ALT=Allele1,AF=Freq1,ES=Effect,SE=StdErr,LP=-logP,SS=N))
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gwas_stats_hg38 <- rtracklayer::liftOver(gwas_granges, chain) %>%
unlist() %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames,
position = start, REF, ALT, AF, ES, SE, LP, SS) %>%
dplyr::mutate(id = paste(chromosome, position, sep = ":")) %>%
dplyr::mutate(MAF = pmin(AF, 1-AF)) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub("chr","",chromosome))
}
microarray <- function(gwas_stats_hg38,ensGene,region38)
{
cat("a. eQTL datasets\n")
microarray_df <- dplyr::filter(tabix_paths, quant_method == "microarray") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(microarray_df$ftp_path), microarray_df$qtl_id[1])
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.CEDAR")
column_names <- names(read.delim(hdr))
summary_list <- purrr::map(ftp_path_list, ~import_eQTLCatalogue(., region38,
selected_gene_id = ensGene, column_names))
purrr::map_df(summary_list[lapply(summary_list,nrow)!=0],
~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
rnaseq <- function(gwas_stats_hg38,ensGene,region38)
{
cat("b. Uniformly processed RNA-seq datasets\n")
rnaseq_df <- dplyr::filter(tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(rnaseq_df$ftp_path), rnaseq_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list, ~safe_import(., region38,
selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list[lapply(result_list,nrow)!=0], ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
purrr::map_df(result_list, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
gtex <- function(gwas_stats_hg38,ensGene,region38)
{
cat("c. GTEx_v8 imported eQTL datasets\n")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_gtex.tsv")
imported_tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>%
dplyr::mutate(ftp_path=file.path("~/rds/public_databases/GTEx/csv",basename(ftp_path)))
gtex_df <- dplyr::filter(imported_tabix_paths, quant_method == "ge") %>%
dplyr::mutate(qtl_id = paste(study, qtl_group, sep = "_"))
ftp_path_list <- setNames(as.list(gtex_df$ftp_path), gtex_df$qtl_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.GTEx")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
prot <- sentinels[["prot"]][r]
invisible(sapply(1:length(result_filtered), function(i) {
if (!is.null(result_filtered[[i]])) {
prot_name <- names(result_filtered)[i]
if (!is.null(prot_name)) {
f <- file.path(analysis, "coloc", "GTEx", "sumstats", paste0(prot, "-", prot_name, ".gz"))
gz <- gzfile(f, "w")
write.table(result_filtered[[i]], file = gz, col.names = TRUE, row.names = FALSE, quote = FALSE, sep = "\t")
close(gz)
} else {
warning(paste("Missing name for index", i))
}
}
}))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "qtl_id")
}
ge <- function(gwas_stats_hg38,ensGene,region38)
{
cat("d. eQTL datasets\n")
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths_ge.tsv")
imported_tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>%
dplyr::mutate(ftp_path=file.path("~/rds/public_databases/eQTLCatalogue",basename(ftp_path)))
ftp_path_list <- setNames(as.list(imported_tabix_paths$ftp_path), imported_tabix_paths$unique_id)
hdr <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","column_names.Alasoo")
column_names <- names(read.delim(hdr))
safe_import <- purrr::safely(import_eQTLCatalogue)
summary_list <- purrr::map(ftp_path_list,
~safe_import(., region38, selected_gene_id = ensGene, column_names))
result_list <- purrr::map(summary_list, ~.$result)
result_list <- result_list[!unlist(purrr::map(result_list, is.null))]
result_filtered <- purrr::map(result_list[lapply(result_list,nrow)!=0],
~dplyr::filter(., !is.na(se)))
prot <- sentinels[["prot"]][r]
invisible(sapply(1:length(result_filtered), function(i) {
if (!is.null(result_filtered[[i]])) {
prot_name <- names(result_filtered)[i]
if (!is.null(prot_name)) {
f <- file.path(analysis, "coloc", "eQTLCatalogue", "sumstats", paste0(prot, "-", prot_name, ".gz"))
gz <- gzfile(f, "w")
write.table(result_filtered[[i]], file = gz, col.names = TRUE, row.names = FALSE, quote = FALSE, sep = "\t")
close(gz)
} else {
warning(paste("Missing name for index", i))
}
}
}))
purrr::map_df(result_filtered, ~run_coloc(., gwas_stats_hg38), .id = "unique_id")
}
gtex_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37,chain)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
if (!exists("df_gtex")) return
saveRDS(df_gtex,file=paste0(out,".rds"))
p <- ggplot(df_gtex, aes(x = PP.H4.abf)) + geom_histogram()
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggplot2::ggsave(plot = s, filename = paste0(out, ".assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggplot2::ggsave(plot = p, filename = paste0(out, ".hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
ge_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_ge <- ge(gwas_stats_hg38,ensGene,region38)
if (!exists("df_ge")) return
saveRDS(df_ge,file=paste0(out,".rds"))
p <- ggplot(df_ge, aes(x = PP.H4.abf)) + geom_histogram()
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, ".assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, ".hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
all_coloc <- function(prot,chr,ensGene,chain,region37,region38,out)
{
gwas_stats_hg38 <- sumstats(prot,chr,region37)
df_microarray <- microarray(gwas_stats_hg38,ensGene,region38)
df_rnaseq <- rnaseq(gwas_stats_hg38,ensGene,region38)
df_gtex <- gtex(gwas_stats_hg38,ensGene,region38)
df_ge <- ge(gwas_stats_hg38,ensGene,region38)
if (exists("df_microarray") & exits("df_rnaseq") & exists("df_gtex") & exists("df_ge"))
{
coloc_df = dplyr::bind_rows(df_microarray, df_rnaseq, df_gtex, df_ge)
saveRDS(coloc_df, file=paste0(out,"-all.rds"))
p <- ggplot(coloc_df, aes(x = PP.H4.abf)) + geom_histogram()
}
s <- ggplot(gwas_stats_hg38, aes(x = position, y = LP)) + geom_point()
ggsave(plot = s, filename = paste0(out, "-assoc.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
ggsave(plot = p, filename = paste0(out, "-hist.pdf"), device = "pdf",
height = 15, width = 15, units = "cm", dpi = 300)
}
single_run <- function(r, batch="GTEx")
{
sentinel <- sentinels[r,]
prot <- sentinel[["prot"]]
chr <- sentinel[["geneChrom"]]
ensRegion37 <- with(sentinel,
{
start <- geneStart-M
if (start<0) start <- 0
end <- geneEnd+M
paste0(chr,":",start,"-",end)
})
ss <- subset(pQTLdata::caprion,Protein==paste0(sentinel[["prot"]],"_HUMAN"))
ensGene <- ss[["ensGenes"]]
x <- with(sentinel,list(chr=geneChrom,start=geneStart,end=geneEnd))
lr <- liftRegion(x)
ensRegion38 <- with(lr,paste0(chr,":",start-M,"-",end+M))
cat(chr,ensGene,ensRegion37,ensRegion38,"\n")
f <- file.path(analysis,"coloc",batch,with(sentinel,paste0(prot,"-",SNP)))
if (batch=="GTEx")
{
gtex_coloc(prot,chr,ensGene,chain,ensRegion37,ensRegion38,f)
} else {
ge_coloc(prot,chr,ensGene,chain,ensRegion37,ensRegion38,f)
}
}
collect <- function(batch="GTEx")
# to collect results when all single runs are done
{
df_coloc <- data.frame()
for(r in 1:nrow(sentinels))
{
prot <- sentinels[["prot"]][r]
snpid <- sentinels[["SNP"]][r]
rsid <- prot_rsid[["SNP"]][r]
f <- file.path(analysis,"coloc",batch,paste0(prot,"-",snpid,".rds"))
if (!file.exists(f)) next
cat(prot,"-",rsid,"\n")
rds <- readRDS(f)
if (nrow(rds)==0) next
df_coloc <- rbind(df_coloc,data.frame(prot=prot,rsid=rsid,snpid=snpid,rds))
}
f <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","hg19ToHg38.over.chain")
chain <- rtracklayer::import.chain(f)
gr <- with(arrange(pQTLdata::caprion,Protein), {
GenomicRanges::GRanges(seqnames = chr,
ranges = IRanges::IRanges(start = start - M, end = end + M),
Protein=Protein,Gene=Gene)
})
seqlevelsStyle(gr) <- "UCSC"
gr38 <- rtracklayer::liftOver(gr,chain)
gr38_new <- lapply(gr38, function(gr) {
valid_start <- start(gr)[!is.na(start(gr))]
valid_end <- end(gr)[!is.na(end(gr))]
if (length(valid_start) > 0 && length(valid_end) > 0) {
min_start <- min(valid_start)
min_end <- min(valid_end)
new_gr <- GRanges(
seqnames = seqnames(gr)[1],
ranges = IRanges(start = min_start, end = min_end),
strand = strand(gr)[1],
Protein = ifelse(length(mcols(gr)$Protein) > 0, unique(mcols(gr)$Protein)[1], NA),
Gene = ifelse(length(mcols(gr)$Gene) > 0, unique(mcols(gr)$Gene)[1], NA)
)
return(new_gr)
} else {
return(NULL)
}
})
gr38_new <- gr38_new[!sapply(gr38_new, is.null)]
df38 <- do.call(rbind, lapply(gr38_new, function(gr) {
data.frame(
seqnames = as.character(seqnames(gr)),
start = start(gr),
end = end(gr),
strand = as.character(strand(gr)),
Protein = mcols(gr)$Protein,
Gene = mcols(gr)$Gene
)
})) %>%
mutate(range38=paste0(gsub("chr","",seqnames),":",start,"-",end)) %>%
select(Protein,Gene,range38)
caprion_upd <- pQTLdata::caprion %>%
mutate(prot=gsub("_HUMAN","",Protein),gene=Gene) %>%
left_join(df38)
df <- dplyr::rename(df_coloc,H0=PP.H0.abf,H1=PP.H1.abf,H2=PP.H2.abf,H3=PP.H3.abf,H4=PP.H4.abf) %>%
dplyr::left_join(caprion_upd[c("prot","gene","range38")])
if (batch=="GTEx") {
df_coloc <- within(df,{qtl_id <- gsub("GTEx_V8_","",qtl_id)})
write.table(subset(df,H4>=0.8),file=file.path(analysis,"coloc","GTEx.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
write.table(df,file=file.path(analysis,"coloc","GTEx-all.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
coloc <- merge(df_coloc,caprion_upd[c("prot","gene","range38")]) %>%
mutate(prot,
H0=round(H0,2),
H1=round(H1,2),
H2=round(H2,2),
H3=round(H3,2),
H4=round(H4,2)) %>%
setNames(c("Protein","Gene","RSid","SNPid","Tissue","nSNP","H0","H1","H2","H3","H4")) %>%
select(Protein,Gene,RSid,Tissue,nSNP,H0,H1,H2,H3,H4)
write.table(coloc,file=file.path(analysis,"coloc","GTEx-ST.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
} else {
write.table(subset(df,H4>=0.8),file=file.path(analysis,"coloc","eQTLCatalogue.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
write.table(df,file=file.path(analysis,"coloc","eQTLCatalogue-all.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
eQTLCatalogue <- left_join(df,caprion_upd[c("prot","gene","range38")]) %>%
mutate(prot,
H0=round(H0,2),
H1=round(H1,2),
H2=round(H2,2),
H3=round(H3,2),
H4=round(H4,2)) %>%
setNames(c("Protein","Gene","RSid","SNPid","Study","nSNP","H0","H1","H2","H3","H4")) %>%
select(Protein,Gene,RSid,Study,nSNP,H0,H1,H2,H3,H4)
write.table(eQTLCatalogue,file=file.path(analysis,"coloc","eQTLCatalogue-ST.tsv"),
quote=FALSE,row.names=FALSE,sep="\t")
}
}
loop_slowly <- function() for (r in 1:nrow(sentinels)) single_run(r)
# Environmental variables
pkgs <- c("dplyr", "gap", "ggplot2", "readr", "coloc", "GenomicRanges","pQTLtools","rtracklayer","seqminer")
invisible(suppressMessages(lapply(pkgs, require, character.only = TRUE)))
options(width=200)
HOME <- Sys.getenv("HOME")
HPC_WORK <- Sys.getenv("HPC_WORK")
analysis <- Sys.getenv("analysis")
M <- 1e6
psum <- file.path(analysis,"coloc","sumstats")
if (!dir.exists(psum)) dir.create(psum)
gsum <- file.path(analysis,"coloc","GTEx","sumstats")
if (!dir.exists(gsum)) dir.create(gsum)
esum <- file.path(analysis,"coloc","eQTLCatalogue","sumstats")
if (!dir.exists(esum)) dir.create(esum)
sevens <- "
ENSG00000131142 - CCL25 19 8052318 8062660
ENSG00000125735 - TNFSF14 19 6661253 6670588
ENSG00000275302 - CCL4 17 36103827 36105621
ENSG00000274736 - CCL23 17 36013056 36017972
ENSG00000013725 - CD6 11 60971680 61020377
ENSG00000138675 - FGF5 4 80266639 80336680
ENSG00000277632 - CCL3 17 36088256 36090169
"
updates <- as.data.frame(scan(file=textConnection(sevens),what=list("","","",0,0,0))) %>%
setNames(c("ensGenes","dash","gene","chromosome","start38","end38"))
caprion <- left_join(pQTLdata::caprion,updates)
sentinels <- subset(read.csv(file.path(analysis,"work","caprion_dr.cis.vs.trans")),cis)
f <- file.path(analysis,"work","snpid_dr.lst")
prot_rsid <- select(sentinels,prot,SNP) %>%
dplyr::left_join(read.table(f,header=TRUE),by=c('SNP'='snpid')) %>%
transmute(prot,SNP=dplyr::if_else(is.na(rsid)|rsid==".",SNP,rsid))
fp <- file.path(find.package("pQTLtools"),"eQTL-Catalogue","tabix_ftp_paths.tsv")
tabix_paths <- read.delim(fp, stringsAsFactors = FALSE) %>% dplyr::as_tibble()
r <- as.integer(Sys.getenv("r"))
single_run(r)
single_run(r,batch="eQTLCatalogue")
collect()
collect(batch="eQTLCatalogue")The single_run() is called for analysis with SLURM and collect() for summary with no need of SLURM.
2.2 LocusZoom.js
This actually involves liftOver back to GRCh37 done as follows for GTEx data.
awk 'NR>1{print $1,$2,$3,$4}' ${analysis}/coloc/GTEx.tsv | \
parallel -C ' ' '
export prot={1}
export rsid={2}
export snpid={3}
export tissue={4}
Rscript -e "
suppressMessages(library(dplyr))
suppressMessages(library(jsonlite))
suppressMessages(library(rtracklayer))
analysis <- Sys.getenv(\"analysis\")
hpc_work <- Sys.getenv(\"HPC_WORK\")
prot <- Sys.getenv(\"prot\")
rsid <- Sys.getenv(\"rsid\")
snpid <- Sys.getenv(\"snpid\")
tissue <- Sys.getenv(\"tissue\")
print(paste0(prot,\"-\",tissue))
f <- file.path(hpc_work,\"bin\",\"hg38ToHg19.over.chain\")
chain <- rtracklayer::import.chain(f)
liftOver <- function(gwas_stats)
{
gwas_granges <- with(gwas_stats,GRanges(seqnames = paste0(\"chr\",dplyr::if_else(chromosome==23,\"X\",paste(chromosome))),
ranges = IRanges(start = position, end = position),
chromosome = chromosome, position = position,
ref_allele=ref_allele,alt_allele=alt_allele,alt_allele_freq=alt_allele_freq,
log_pvalue=log_pvalue,beta=beta,se=se))
gwas_stats_hg37 <- rtracklayer::liftOver(gwas_granges, chain) %>%
unlist() %>%
dplyr::as_tibble() %>%
dplyr::transmute(chromosome = seqnames, position = start,
variant = paste0(chromosome,\":\",position,\"_\",ref_allele,\"/\",alt_allele),
ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
dplyr::mutate(id = paste(chromosome, position, sep = \":\")) %>%
dplyr::group_by(id) %>%
dplyr::mutate(row_count = n()) %>%
dplyr::ungroup() %>%
dplyr::filter(row_count == 1) %>%
mutate(chromosome=gsub(\"chr\",\"\",chromosome))
}
pGWAS_sumstats <- read.delim(file.path(analysis,\"coloc\",\"sumstats\",paste0(prot,\"-\",snpid,\".gz\"))) %>%
dplyr::mutate(REF=toupper(REF),ALT=toupper(ALT),variant=paste0(chromosome,\":\",position,\"_\",REF,\"/\",ALT)) %>%
dplyr::mutate(log_pvalue=LP,ref_allele=REF,alt_allele=ALT,alt_allele_freq=AF,beta=ES,se=SE) %>%
dplyr::select(chromosome,position,variant,ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
liftOver()
GTEx_sumstats <- read.delim(file.path(analysis,\"coloc\",\"GTEx\",\"sumstats\",paste0(prot,\"-\",tissue,\".gz\"))) %>%
dplyr::mutate(REF=toupper(ref),ALT=toupper(alt),variant=paste0(chromosome,\":\",position,\"_\",REF,\"/\",ALT)) %>%
dplyr::mutate(log_pvalue=-log10(pvalue),ref_allele=REF,alt_allele=ALT,alt_allele_freq=ac/an) %>%
dplyr::select(chromosome,position,variant,ref_allele,alt_allele,alt_allele_freq,log_pvalue,beta,se) %>%
liftOver()
j <- gzfile(file.path(analysis,\"json\",\"pqtleqtl\",paste0(prot,\"-\",tissue,\".json.gz\")))
combined_json <- jsonlite::toJSON(list(pqtl=pGWAS_sumstats,eqtl=GTEx_sumstats),auto_unbox=TRUE,pretty=TRUE)
write(combined_json,file=j)
close(j)
"
'Note that compressed json (.json.gz) is used.